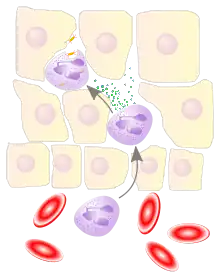
Leukocyte extravasation (also commonly known as leukocyte adhesion cascade or diapedesis – the passage of cells through the intact vessel wall) is the movement of leukocytes out of the circulatory system and towards the site of tissue damage or infection. This process forms part of the innate immune response, involving the recruitment of non-specific leukocytes. Monocytes also use this process in the absence of infection or tissue damage during their development into macrophages.
Overview
Leukocyte extravasation occurs mainly in post-capillary venules, where haemodynamic shear forces are minimised. This process can be understood in several steps:
- Chemoattraction
- Rolling adhesion
- Tight adhesion
- (Endothelial) Transmigration
It has been demonstrated that leukocyte recruitment is halted whenever any of these steps is suppressed.
White blood cells (leukocytes) perform most of their functions in tissues. Functions include phagocytosis of foreign particles, production of antibodies, secretion of inflammatory response triggers (histamine and heparin), and neutralization of histamine. In general, leukocytes are involved in the defense of an organism and protect it from disease by promoting or inhibiting inflammatory responses. Leukocytes use the blood as a transport medium to reach the tissues of the body. Here is a brief summary of each of the four steps currently thought to be involved in leukocyte extravasation:
Chemoattraction
Upon recognition of and activation by pathogens, resident macrophages in the affected tissue release cytokines such as IL-1, TNFα and chemokines. IL-1, TNFα and C5a[1] cause the endothelial cells of blood vessels near the site of infection to express cellular adhesion molecules, including selectins. Circulating leukocytes are localised towards the site of injury or infection due to the presence of chemokines.
Rolling adhesion
Like velcro, carbohydrate ligands on the circulating leukocytes bind to selectin molecules on the inner wall of the vessel, with marginal affinity. This causes the leukocytes to slow down and begin rolling along the inner surface of the vessel wall. During this rolling motion, transitory bonds are formed and broken between selectins and their ligands.
For example, the carbohydrate ligand for P-selectin, P-selectin glycoprotein ligand-1 (PSGL-1), is expressed by different types of leukocytes (white blood cells). The binding of PSGL-1 on the leukocyte to P-selectin on the endothelial cell allows for the leukocyte to roll along the endothelial surface. This interaction can be tuned by the glycosylation pattern of PSGL-1, such that certain glycovariants of PSGL-1 will have unique affinities for different selectins, allowing in some cases for cells to migrate to specific sites within the body (e.g. the skin).[2]
Tight adhesion
At the same time, chemokines released by macrophages activate the rolling leukocytes and cause surface integrin molecules to switch from the default low-affinity state to a high-affinity state. This is assisted through juxtacrine activation of integrins by chemokines and soluble factors released by endothelial cells. In the activated state, integrins bind tightly to complementary receptors expressed on endothelial cells, with high affinity. This causes the immobilization of the leukocytes, which varies in vessels that contain different shear forces of the ongoing blood flow.
Transmigration
The cytoskeletons of the leukocytes are reorganized in such a way that the leukocytes are spread out over the endothelial cells. In this form, leukocytes extend pseudopodia and pass through gaps between endothelial cells. This passage of cells through the intact vessel wall is called diapedesis.[3] These gaps can form through interactions of the leukocytes with the endothelium, but also autonomously through endothelial mechanics.[4] Transmigration of the leukocyte occurs as PECAM proteins, found on the leukocyte and endothelial cell surfaces, interact and effectively pull the cell through the endothelium. Once through the endothelium, the leukocyte must penetrate the basement membrane. The mechanism for penetration is disputed, but may involve proteolytic digestion of the membrane, mechanical force, or both.[5] The entire process of blood vessel escape is known as diapedesis. Once in the interstitial fluid, leukocytes migrate along a chemotactic gradient towards the site of injury or infection.
Molecular biology
Introduction
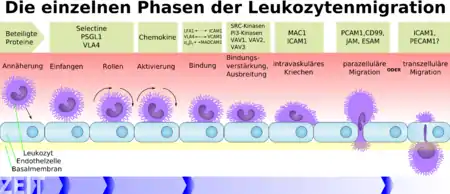
The phases of the leukocyte extravasation depicted in the schema are: approach, capture, rolling, activation, binding, strengthening of the binding and spreading, intravascular creeping, paracellular migration or transcellular migration.
Selectins
Selectins are expressed shortly after cytokine activation of endothelial cells by tissue macrophages. Activated endothelial cells initially express P-selectin molecules, but within two hours after activation E-selectin expression is favoured. Endothelial selectins bind carbohydrates on leukocyte transmembrane glycoproteins, including sialyl-LewisX.
- P-selectins: P-selectin is expressed on activated endothelial cells and platelets. Synthesis of P-selectin can be induced by thrombin, leukotriene B4, complement fragment C5a, histamine, TNFα or LPS. These cytokines induce the externalisation of Weibel-Palade bodies in endothelial cells, presenting pre-formed P-selectins on the endothelial cell surface. P-selectins bind PSGL-1 as a ligand.[6]
- E-selectins: E-selectin is expressed on activated endothelial cells. Synthesis of E-selectin follows shortly after P-selectin synthesis, induced by cytokines such as IL-1 and TNFα. E-selectins bind PSGL-1 and ESL-1.
- L-selectins: L-selectins are constitutively expressed on some leukocytes, and are known to bind GlyCAM-1, MadCAM-1 and CD34 as ligands.
Suppressed expression of some selectins results in a slower immune response. If L-selectin is not produced, the immune response may be ten times slower, as P-selectins (which can also be produced by leukocytes) bind to each other. P-selectins can bind each other with high affinity, but occur less frequently because the receptor-site density is lower than with the smaller E-selectin molecules. This increases the initial leukocyte rolling speed, prolonging the slow rolling phase.
Integrins
Integrins involved in cellular adhesion are primarily expressed on leukocytes. β2 integrins on rolling leukocytes bind endothelial cellular adhesion molecules, arresting cell movement.
- LFA-1 is found on circulating leukocytes, and binds ICAM-1 and ICAM-2 on endothelial cells
- Mac-1 is found on circulating leukocytes, and binds ICAM-1 on endothelial cells
- VLA-4 is found on leukocytes and endothelial cells, and facilitates chemotaxis; it also binds VCAM-1
Cellular activation via extracellular chemokines causes pre-formed β2 integrins to be released from cellular stores. Integrin molecules migrate to the cell surface and congregate in high-avidity patches. Intracellular integrin domains associate with the leukocyte cytoskeleton, via mediation with cytosolic factors such as talin, α-actinin and vinculin. This association causes a conformational shift in the integrin's tertiary structure, allowing ligand access to the binding site. Divalent cations (e.g. Mg2+) are also required for integrin-ligand binding.
Integrin ligands ICAM-1 and VCAM-1 are activated by inflammatory cytokines, while ICAM-2 is constitutively expressed by some endothelial cells but downregulated by inflammatory cytokines. ICAM-1 and ICAM-2 share two homologous N-terminal domains; both can bind LFA-1.
During chemotaxis, cell movement is facilitated by the binding of β1 integrins to components of the extracellular matrix: VLA-3, VLA-4 and VLA-5 to fibronectin and VLA-2 and VLA-3 to collagen and other extracellular matrix components.
Cytokines
Extravasation is regulated by the background cytokine environment produced by the inflammatory response, and is independent of specific cellular antigens. Cytokines released in the initial immune response induce vasodilation and lower the electrical charge along the vessel's surface. Blood flow is slowed, facilitating intermolecular binding.
- IL-1 activates resident lymphocytes and vascular endothelia
- TNFα increases vascular permeability and activates vascular endothelia
- CXCL8 (IL-8) forms a chemotactic gradient that directs leukocytes towards site of tissue injury/infection (CCL2 has a similar function to CXCL8, inducing monocyte extravasation and development into macrophages); also activates leukocyte integrins
Recent advances
In 1976, SEM images showed that there were homing receptors on microvilli-like tips on leukocytes that would allow white blood cells to get out of the blood vessel and get into tissue.[7] Since the 1990s the identity of ligands involved in leukocyte extravasation have been studied heavily. This topic was finally able to be studied thoroughly under physiological shear stress conditions using a typical flow chamber.[8] Since the first experiments, a strange phenomenon was observed. Binding interactions between the white blood cells and the vessel walls were observed to become stronger under higher force. Selectins (E-selectin, L-selectin, and P-selectin) were found to be involved in this phenomenon. The shear threshold requirement seems counterintuitive because increasing shear elevates the force applied to adhesive bonds and it would seem that this should increase the dislodging ability. Nevertheless, cells roll more slowly and more regularly until an optimal shear is reached where rolling velocity is minimal. This paradoxical phenomenon has not been satisfactorily explained despite the widespread interest.
One initially dismissed hypothesis that has been gaining interest is the catch bond hypothesis, where the increased force on the cell slows off-rates and lengthen the bond lifetimes and stabilizing the rolling step of leukocyte extravasation.[9] Flow-enhanced cell adhesion is still an unexplained phenomenon that could result from a transport-dependent increase in on-rates or a force-dependent decrease in off-rates of adhesive bonds. L-selectin requires a particular minimum of shear to sustain leukocyte rolling on P-selectin glycoprotein ligand-1 (PSGL-1) and other vascular ligands. It has been hypothesized that low forces decrease L-selectin–PSGL-1 off-rates (catch bonds), whereas higher forces increase off-rates (slip bonds). Experiments have found that a force-dependent decrease in off-rates dictated flow-enhanced rolling of L-selectin–bearing microspheres or neutrophils on PSGL-1. [5] Catch bonds enable increasing force to convert short bond lifetimes into long bond lifetimes, which decrease rolling velocities and increase the regularity of rolling steps as shear rose from the threshold to an optimal value. As shear increases, transitions to slip bonds shorten their bond lifetimes and increase rolling velocities and decrease rolling regularity. It is hypothesized that force-dependent alterations of bond lifetimes govern L-selectin–dependent cell adhesion below and above the shear optimum. These findings establish a biological function for catch bonds as a mechanism for flow-enhanced cell adhesion.[10] While leukocytes seem to undergo a catch bond behavior with increasing flow leading to the tethering and rolling steps in leukocyte extravasation, firm adhesion is achieved through another mechanism, integrin activation.
Other biological examples of a catch bond mechanism is seen in bacteria that tightly cling to urinary tract walls in response to high fluid velocities and large shear forces exerted on the cells and bacteria with adhesive tips of fimbria.[9][11] Schematic mechanisms of how increased shear force is proposed to cause stronger binding interactions between bacteria and target cells show that the catch bond acts very similar to a Chinese finger trap. For a catch-bond, the force on the cell pulls the adhesive tip of a fimbria to close tighter on its target cell. As the strength of the forces increases, the stronger the bond between the fimbria and the cell-receptor on the surface of the target cell.[11] For a cryptic-bond, the force causes the fimbria to swivel toward the target cell and have more binding sites able to attach to the target cell ligands, mainly sugar molecules. This creates a stronger bonding interaction between the bacteria and the target cell.
Advent of microfluidic devices
Parallel plate flow chambers are among the most popular flow chambers used to study the leukocyte-endothelial interaction in vitro. They have been used for investigation since the later 1980s.[12] Although flow chambers have been an important tool to study leukocyte rolling, there are several limitations when it comes to studying the physiological in vivo conditions, as they lack correspondence with in vivo geometry, including scale/aspect ratio (microvasculature vs large vessel models), flow conditions (e.g. converging vs diverging flows at bifurcations), and require large reagent volumes (~ ml) due to their large size (height > 250 µm and width > 1mm).[13] With the advent of microfluidic-based devices, these limitations have been overcome. A new in vitro model, called SynVivo Synthetic microvascular network (SMN) was produced by the CFD Research Corporation (CFDRC) and developed using the polydimethylsiloxane (PDMS) based soft-lithography process. The SMN can recreate the complex in vivo vasculature, including geometrical features, flow conditions, and reagent volumes, thereby providing a biologically realistic environment for studying the extravasation cellular behavior, but also for drug delivery and drug discovery. [14][15]
Leukocyte adhesion deficiency
Leukocyte adhesion deficiency (LAD) is a genetic disease associated with a defect in the leukocyte extravasation process, caused by a defective integrin β2 chain (found in LFA-1 and Mac-1). This impairs the ability of the leukocytes to stop and undergo diapedesis. People with LAD suffer from recurrent bacterial infections and impaired wound healing. Neutrophilia is a hallmark of LAD.
Neutrophil dysfunction
In widespread diseases such as sepsis, leukocyte extravasation enters an uncontrolled stage, where white blood neutrophils begin destroying host tissues at unprecedented rates, claiming the lives of about 200,000 people in the United States alone.[16] Neutrophil dysfunction is usually preceded by an infection of some sort, which triggers pathogen-associated molecular patterns (PAMP). As leukocyte extravasation intensifies, more tissues are damaged by neutrophils, which release oxygen radicals and proteases.[16]
Recent studies with SynVivo Synthetic microvascular network (SMN) made it possible to study anti-inflammatory therapeutics to treat pathologies caused by neutrophil dysfunction. The SMN enables the thorough analysis of each stage of leukocyte extravasation, thereby providing a methodology to quantify the effect of the drug in impeding leukocyte extravasation. Some of the recent findings demonstrate the effect of hydrodynamics on neutrophil-endothelial interactions. In other words, adhesion of neutrophils is heavily impacted by shear forces as well as molecular interactions. Moreover, as shear rate decreases (e.g., in post-capillary venules), immobilization of the leukocytes becomes easier and thus, more prevalent. The opposite is also true; vessels in which shear forces are high render the immobilization of the leukocytes more difficult. This has high implications in various diseases, where disruptions in blood flow gravely impact immune system response by impeding or expediting the immobilization of the leukocytes. Having this knowledge allows for better studies of the effect of drugs on leukocyte extravasation.[13][16][14]
Footnotes
- ↑ Monk PN, Scola AM, Madala P, Fairlie DP (October 2007). "Function, structure and therapeutic potential of complement C5a receptors". British Journal of Pharmacology. 152 (4): 429–48. doi:10.1038/sj.bjp.0707332. PMC 2050825. PMID 17603557.
- ↑ Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB (February 2015). "Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review". Journal of Autoimmunity. 57 (6): 1–13. doi:10.1016/j.jaut.2014.12.002. PMC 4340844. PMID 25578468.
- ↑ Beekhuizen, Henry; Furth, Ralph van (1998). "Diapedesis". Encyclopedia of Immunology. pp. 757–760. doi:10.1006/rwei.1999.0200. ISBN 978-0-12-226765-9.
- ↑ Escribano J, Chen MB, Moeendarbary E, Cao X, Shenoy V, Garcia-Aznar JM, et al. (May 2019). "Balance of mechanical forces drives endothelial gap formation and may facilitate cancer and immune-cell extravasation". PLOS Computational Biology. 15 (5): e1006395. arXiv:1811.09326. Bibcode:2019PLSCB..15E6395E. doi:10.1371/journal.pcbi.1006395. PMC 6497229. PMID 31048903.
- ↑ Sorokin L (October 2010). "The impact of the extracellular matrix on inflammation". Nature Reviews. Immunology. Nature Publishing Group. 10 (10): 712–23. doi:10.1038/nri2852. PMID 20865019. S2CID 18130467.
- ↑ McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF (July 1989). "GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies". The Journal of Clinical Investigation. 84 (1): 92–9. doi:10.1172/JCI114175. PMC 303957. PMID 2472431.
- ↑ Anderson AO, Anderson ND (November 1976). "Lymphocyte emigration from high endothelial venules in rat lymph nodes". Immunology. 31 (5): 731–48. PMC 1445135. PMID 992709.
- ↑ Wiese G, Barthel SR, Dimitroff CJ (February 2009). "Analysis of physiologic E-selectin-mediated leukocyte rolling on microvascular endothelium". Journal of Visualized Experiments. 24 (24): 1009. doi:10.3791/1009. PMC 2730781. PMID 19229187.
- 1 2 Thomas WE, Nilsson LM, Forero M, Sokurenko EV, Vogel V (September 2004). "Shear-dependent 'stick-and-roll' adhesion of type 1 fimbriated Escherichia coli". Molecular Microbiology. 53 (5): 1545–57. doi:10.1111/j.1365-2958.2004.04226.x. PMID 15387828. S2CID 24777923.
- ↑ Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP (September 2004). "Catch bonds govern adhesion through L-selectin at threshold shear". The Journal of Cell Biology. 166 (6): 913–23. doi:10.1083/jcb.200403144. PMC 2172126. PMID 15364963.
- 1 2 Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV (June 2002). "Bacterial adhesion to target cells enhanced by shear force". Cell. 109 (7): 913–23. doi:10.1016/S0092-8674(02)00796-1. PMID 12110187.
- ↑ Nabel G, Baltimore D (1987). "An inducible transcription factor activates expression of human immunodeficiency virus in T cells". Nature. 326 (6114): 711–3. Bibcode:1987Natur.326..711N. doi:10.1038/326711a0. PMID 3031512. S2CID 4317942.
- 1 2 Prabhakarpandian B, Shen MC, Pant K, Kiani MF (November 2011). "Microfluidic devices for modeling cell-cell and particle-cell interactions in the microvasculature". Microvascular Research. 82 (3): 210–20. doi:10.1016/j.mvr.2011.06.013. PMC 3215799. PMID 21763328.
- 1 2 Smith AM, Prabhakarpandian B, Pant K (May 2014). "Generation of shear adhesion map using SynVivo synthetic microvascular networks". Journal of Visualized Experiments. 87 (87): e51025. doi:10.3791/51025. PMC 4207183. PMID 24893648.
- ↑ Lamberti G, Prabhakarpandian B, Garson C, Smith A, Pant K, Wang B, Kiani MF (August 2014). "Bioinspired microfluidic assay for in vitro modeling of leukocyte-endothelium interactions". Analytical Chemistry. 86 (16): 8344–51. doi:10.1021/ac5018716. PMC 4139165. PMID 25135319.
- 1 2 3 Soroush F, Zhang T, King DJ, Tang Y, Deosarkar S, Prabhakarpandian B, Kilpatrick LE, Kiani MF (November 2016). "A novel microfluidic assay reveals a key role for protein kinase C δ in regulating human neutrophil-endothelium interaction". Journal of Leukocyte Biology. 100 (5): 1027–1035. doi:10.1189/jlb.3MA0216-087R. PMC 5069089. PMID 27190303.
References
- Aplin AE, Howe A, Alahari SK, Juliano RL (June 1998). "Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins". Pharmacological Reviews. 50 (2): 197–263. PMID 9647866.
- Anderson AO, Anderson ND (November 1976). "Lymphocyte emigration from high endothelial venules in rat lymph nodes". Immunology. 31 (5): 731–48. PMC 1445135. PMID 992709.
- Wiese G, Barthel SR, Dimitroff CJ (February 2009). "Analysis of physiologic E-selectin-mediated leukocyte rolling on microvascular endothelium". Journal of Visualized Experiments. 24 (24): 1009. doi:10.3791/1009. PMC 2730781. PMID 19229187.
- Thomas WE, Nilsson LM, Forero M, Sokurenko EV, Vogel V (September 2004). "Shear-dependent 'stick-and-roll' adhesion of type 1 fimbriated Escherichia coli". Molecular Microbiology. 53 (5): 1545–57. doi:10.1111/j.1365-2958.2004.04226.x. PMID 15387828. S2CID 24777923.
- Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP (September 2004). "Catch bonds govern adhesion through L-selectin at threshold shear". The Journal of Cell Biology. 166 (6): 913–23. doi:10.1083/jcb.200403144. PMC 2172126. PMID 15364963.
- Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV (June 2002). "Bacterial adhesion to target cells enhanced by shear force". Cell. 109 (7): 913–23. doi:10.1016/S0092-8674(02)00796-1. PMID 12110187.
- Dept of Biomedical Engineering, University of Virginia. "Inflammation: The Leukocyte Adhesion Cascade".
- Janeway CA, Travers P, Walport M, Shlomick MJ (2005). Immunobiology: The immune system in health and disease (6th ed.). New York: Garland Science Publishing. pp. 76–84. ISBN 0-8153-4101-6.
- Kindt TJ, Osborne BA, Goldsby RA (2007). "Overview of the Immune System". Kuby Immunology (6th ed.).
- Mak TW and Saunders ME; Chaddah MR; Tamminen WL (2006). The Immune Response: Basic and Clinical Principles. Oxford, UK: Elsevier Inc. pp. 61–2.
- Male D, Brostoff J, Roth DB, Roitt I (2006). Immunology (7th ed.). Philadelphia: Elsevier Limited. pp. 130–8.
- Nairn R, Helbert M (2002). Immunology for Medical Students. Edinburgh: Harcourt Publishers Ltd. pp. 106–7.
- "Animation of the adhesion process".