In chemistry, transfer hydrogenation is a chemical reaction involving the addition of hydrogen to a compound from a source other than molecular H2. It is applied in laboratory and industrial organic synthesis to saturate organic compounds and reduce ketones to alcohols, and imines to amines. It avoids the need for high-pressure molecular H2 used in conventional hydrogenation. Transfer hydrogenation usually occurs at mild temperature and pressure conditions using organic or organometallic catalysts, many of which are chiral, allowing efficient asymmetric synthesis. It uses hydrogen donor compounds such as formic acid, isopropanol or dihydroanthracene, dehydrogenating them to CO2, acetone, or anthracene respectively.[1] Often, the donor molecules also function as solvents for the reaction. A large scale application of transfer hydrogenation is coal liquefaction using "donor solvents" such as tetralin.[2][3]
Organometallic catalysts
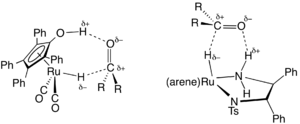
In the area of organic synthesis, a useful family of hydrogen-transfer catalysts have been developed based on ruthenium and rhodium complexes, often with diamine and phosphine ligands.[4] A representative catalyst precursor is derived from (cymene)ruthenium dichloride dimer and the tosylated diphenylethylenediamine. These catalysts are mainly employed for the reduction of ketones and imines to alcohols and amines, respectively. The hydrogen-donor (transfer agent) is typically isopropanol, which converts to acetone upon donation of hydrogen. Transfer hydrogenations can proceed with high enantioselectivities when the starting material is prochiral:

where RR'C*H−OH is a chiral product. A typical catalyst is (cymene)R,R-HNCHPhCHPhNTs, where Ts refers to a tosyl group (SO2C6H4Me) and R,R refers to the absolute configuration of the two chiral carbon centers. This work was recognized with the 2001 Nobel Prize in Chemistry to Ryōji Noyori.[5]
Another family of hydrogen-transfer agents are those based on aluminium alkoxides, such as aluminium isopropoxide in the MPV reduction; however their activities are relatively low by comparison with the transition metal-based systems.
The catalytic asymmetric hydrogenation of ketones was demonstrated with ruthenium-based complexes of BINAP.[6][7]
Even though the BINAP-Ru dihalide catalyst could reduce functionalized ketones, the hydrogenation of simple ketones remained unsolved. This challenge was solved with precatalysts of the type RuCl2(diphosphane)(diamine).[8] These catalysts preferentially reduce ketones and aldehydes, leaving olefins and many other substituents unaffected.

Metal-free routes
Prior to the development of catalytic hydrogenation, many methods were developed for the hydrogenation of unsaturated substrates. Many of these methods are only of historical and pedagogical interest. One prominent transfer hydrogenation agent is diimide or (NH)2, also called diazene. This becomes oxidized to the very stable N2:
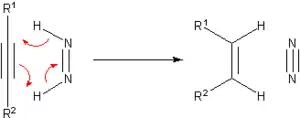
The diimide can be generated from hydrazine or certain other organic precursors.
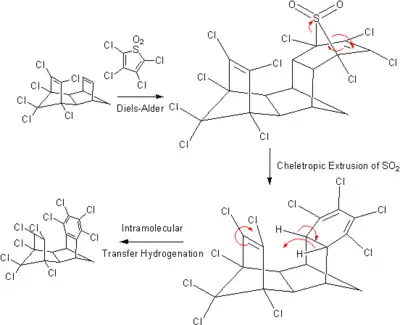
Two hydrocarbons that can serve as hydrogen donors are cyclohexene or cyclohexadiene. In this case, an alkane is formed, along with a benzene. The gain of aromatic stabilization energy when the benzene is formed is the driving force of the reaction. Pd can be used as a catalyst and a temperature of 100 °C is employed. More exotic transfer hydrogenations have been reported, including this intramolecular one:
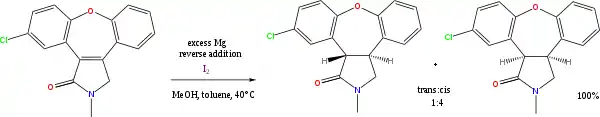
Many reactions exist with alcohol or amines as the proton donors, and alkali metals as electron donors. Of continuing value is the sodium metal-mediated Birch reduction of arenes (another name for aromatic hydrocarbons). Less important presently is the Bouveault–Blanc reduction of esters. The combination of magnesium and methanol is used in alkene reductions, e.g. the synthesis of asenapine:[11]
Organocatalytic transfer hydrogenation
Organocatalytic transfer hydrogenation has been described by the group of List in 2004 in a system with a Hantzsch ester as hydride donor and an amine catalyst:[12]
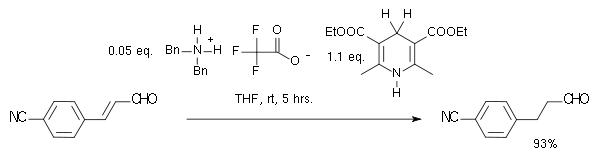
In this particular reaction the substrate is an α,β-unsaturated carbonyl compound. The proton donor is oxidized to the pyridine form and resembles the biochemically relevant coenzyme NADH. In the catalytic cycle for this reaction the amine and the aldehyde first form an iminium ion, then proton transfer is followed by hydrolysis of the iminium bond regenerating the catalyst. By adopting a chiral imidazolidinone MacMillan organocatalyst an enantioselectivity of 81% ee was obtained:
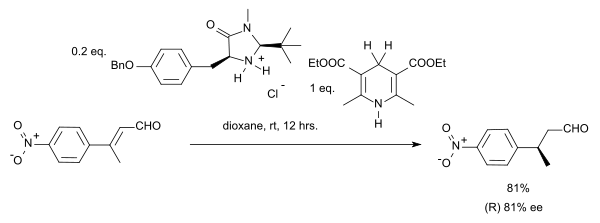
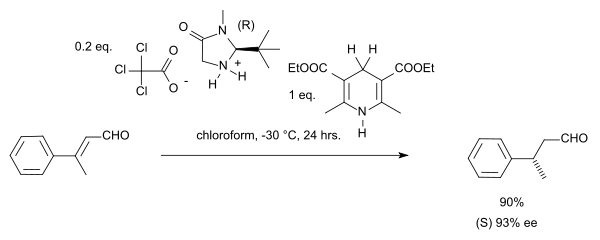
In a case of stereoconvergence, both the E-isomer and the Z-isomer in this reaction yield the (S)-enantiomer.
Extending the scope of this reaction towards ketones or rather enones requires fine tuning of the catalyst (add a benzyl group and replace the t-butyl group by a furan) and of the Hantzsch ester (add more bulky t-butyl groups):[14]
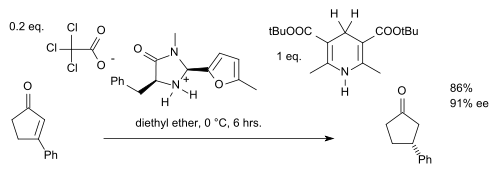
With another organocatalyst altogether, hydrogenation can also be accomplished for imines. One cascade reaction is catalyzed by a chiral phosphoric acid:[15]
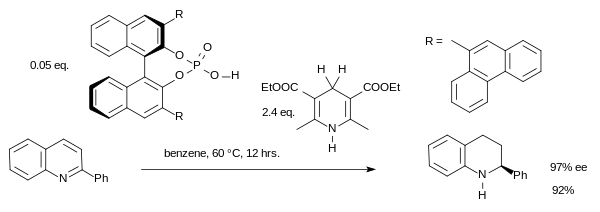
The reaction proceeds via a chiral iminium ion. Traditional metal-based catalysts, hydrogenation of aromatic or heteroaromatic substrates tend to fail.
References
- ↑ Wang, Dong; Astruc, Didier (2015). "The Golden Age of Transfer Hydrogenation". Chem. Rev. 115 (13): 6621–6686. doi:10.1021/acs.chemrev.5b00203. ISSN 0009-2665.
- ↑ Speight, J. G. "The Chemistry and Technology of Coal" Marcel Dekker; New York, 1983; p. 226 ff. ISBN 0-8247-1915-8.
- ↑ Muñiz, Kilian (2005). "Bifunctional Metal-Ligand Catalysis: Hydrogenations and New Reactions within the Metal-(Di)amine Scaffold13". Angewandte Chemie International Edition. 44 (41): 6622–6627. doi:10.1002/anie.200501787. PMID 16187395.
- ↑ T. Ikariya, K. Murata, R. Noyori "Bifunctional Transition Metal-Based Molecular Catalysts for Asymmetric Syntheses" Org. Biomol. Chem., 2006, volume 4, 393-406.
- ↑ Shimizu, H., Nagasaki, I., Matsumura, K., Sayo, N., and Saito, T. "Developments in Asymmetric Hydrogenation from an Industrial Perspective" Acc. Chem. Res. 2007, vol. 40, pp. 1385-1393. doi:10.1021/ar700101x
- ↑ Mashima, K.; Kusano, K.-h.; Sato, N.; Matsumura, Y.-i.; Nozaki, K.; Kumobayashi, H.; Sayo, N.; Hori, Y.; Ishizaki, T. (1994), "Cationic BINAP-Ru(II) Halide Complexes: Highly Efficient Catalysts for Stereoselective Asymmetric Hydrogenation of α- and β-Functionalized Ketones", The Journal of Organic Chemistry, 59 (11): 3064–3076, doi:10.1021/jo00090a026
- ↑ Kitamura, M.; Ohkuma, T.; Inoue, S.; Sayo, N.; Kumobayashi, H.; Akutagawa, S.; Ohta, T.; Takaya, H.; Noyori, R. (1988), "Homogeneous Asymmetric Hydrogenation of functionalized ketones", Journal of the American Chemical Society, 110 (2): 629–631, doi:10.1021/ja00210a070
- ↑ Noyori, R.; Ohkuma, T. (2001), "Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones", Angewandte Chemie International Edition, 40 (1): 40–73, doi:10.1002/1521-3773(20010105)40:1<40::aid-anie40>3.0.co;2-5, PMID 11169691
- ↑ Dub, Pavel A.; Gordon, John C. (2018). "The role of the metal-bound N–H functionality in Noyori-type molecular catalysts". Nature Reviews Chemistry. 2 (12): 396–408. doi:10.1038/s41570-018-0049-z. S2CID 106394152.
- ↑ M. M. Midland (1989). "Asymmetric reductions with organoborane reagents". Chemical Reviews. 89 (7): 1553–1561. doi:10.1021/cr00097a010.
- ↑ Linden, M. V. D.; Roeters, T.; Harting, R.; Stokkingreef, E.; Gelpke, A. S.; Kemperman, G. (2008). "Debottlenecking the Synthesis Route of Asenapine". Organic Process Research & Development. 12 (2): 196–201. doi:10.1021/op700240c.
- ↑ Yang; Hechavarria Fonseca, M.; List, B. (2004). "A metal-free transfer hydrogenation: organocatalytic conjugate reduction of alpha,beta-unsaturated aldehydes". Angewandte Chemie International Edition in English. 43 (48): 6660–6662. doi:10.1002/anie.200461816. PMID 15540245.
- ↑ Ouellet; Tuttle, J.; MacMillan, D. (2005). "Enantioselective organocatalytic hydride reduction". Journal of the American Chemical Society. 127 (1): 32–33. doi:10.1021/ja043834g. PMID 15631434.
- ↑ Tuttle; Ouellet, S.; MacMillan, D. (2006). "Organocatalytic transfer hydrogenation of cyclic enones" (PDF). Journal of the American Chemical Society. 128 (39): 12662–12663. doi:10.1021/ja0653066. PMID 17002356. S2CID 12456921.
- ↑ Rueping; Antonchick, A.; Theissmann, T. (2006). "A highly enantioselective Brønsted acid catalyzed cascade reaction: organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids". Angewandte Chemie International Edition in English. 45 (22): 3683–3686. doi:10.1002/anie.200600191. PMID 16639754.