| Complement deficiency | |
|---|---|
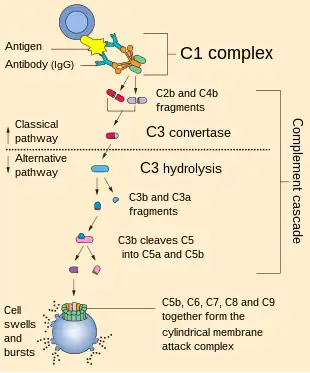 | |
| Complement pathway (normal) | |
| Specialty | Hematology |
| Symptoms | Recurring infection, rheumatic disease[1] |
| Causes | Can be inherited or acquired[2] |
| Diagnostic method | CH50 measurement, Plasma level[3] |
| Treatment | Immunosuppressive therapy[2] |
Complement deficiency is an immunodeficiency of absent or suboptimal functioning of one of the complement system proteins.[4] Because of redundancies in the immune system, many complement disorders are never diagnosed. Some studies estimate that less than 10% are identified.[5] Hypocomplementemia may be used more generally to refer to decreased complement levels,[6] while secondary complement disorder means decreased complement levels that are not directly due to a genetic cause but secondary to another medical condition.[7]
Signs and symptoms
The following symptoms (signs) are consistent with complement deficiency in general:[1][3][8]
- Recurring infection
- Auto-immune disorders
- Glomerulonephritis
- Joint problems (manifestation)
- Lung function (MBL variant alleles)
- Angioedema
- Dermatomyositis
- Vasculitis
- Anaphylactoid purpura
Complications
Vaccinations for encapsulated organisms (e.g., Neisseria meningitidis and Streptococcus pneumoniae) is crucial for preventing infections in complement deficiencies. Among the possible complications are the following:
Causes
The cause of complement deficiency is genetics (though cases of an acquired nature do exist post infection). The majority of complement deficiencies are inherited as autosomal recessive conditions, while properdin deficiency occurs through X-linked inheritance. MBL deficiency can be inherited by either manner.[2]
Inherited
- Properdin deficiency is an X-linked[10] disorder that also causes susceptibility to Neisseria infections.[2]
- C1-inhibitor deficiency or hereditary angioedema will have low C4 with normal C1 levels.[11]
Acquired
Acquired hypocomplementemia may occur in the setting of bone infections (osteomyelitis), infection of the lining of the heart (endocarditis), and cryoglobulinemia. Systemic lupus erythematosus is associated with low C3 and C4.[12] Membranoproliferative glomerulonephritis usually has low C3.[13]
Mechanism

The mechanism of complement deficiency consists of:
- C2: In regard to C2 deficiency, about 5 different mutations in the C2 gene are responsible. In turn, immune function decreases and infection opportunities increase. One of the most common mutations deletes 28 DNA nucleotides from the C2 gene. Therefore, no C2 protein which can help make C3-convertase is produced. Ultimately, this delays/decreases immune response.[14]
- C3: In terms of deficiency of C3, it has been found that 17 mutations in the C3 gene cause problems with C3. This rare condition mutates or prevents C3 protein from forming, lowering the immune system's ability to protect.[15]
- C4: C4 deficiency is highly associated with systemic lupus erythematosus.[3] Aβ42, a protein involved in Alzheimer's disease, can cause activation of C4 (even in plasma deficient of C1q).[16] At least one study indicates that the genetic variation of C4 plays a role in schizophrenia.[17]
Diagnosis
| C4 (C) | FB (A) | C3 | CH50 | Conditions |
|---|---|---|---|---|
| · | ↓ | ↓ | ↓ | PSG, C3 NeF AA |
| ↓ | · | ↓ | · | HAE, C4D |
| · | · | · | ↓ | TCPD |
| ↓ | ·/↓ | ↓ | ↓ | SLE |
| ↑ | ↑ | ↑ | ↑ | inflammation |
The diagnostic tests used to diagnose a complement deficiency include:[3]
- CH50 measurement
- Immunochemical methods/test
- C3 deficiency screening
- Mannose-binding lectin (lab study)
- Plasma levels/regulatory proteins (lab study)
Types
- Disorders of the proteins that act to inhibit the complement system (such as C1-inhibitor) can lead to an overactive response, causing conditions such as hereditary angioedema.[18]
- Disorders of the proteins that act to activate the complement system (such as C3) can lead to an underactive response, causing greater susceptibility to infections.[19]
Treatment
In terms of management for complement deficiency, immunosuppressive therapy should be used depending on the disease presented. A C1-INH concentrate can be used for angio-oedema (C1-INH deficiency).[2][3]
Pneumococcus and Haemophilus infections can be prevented via immunization.[2] Epsilon-aminocaproic acid could be used to treat hereditary C1-INH deficiency, though the possible side effect of intravascular thrombosis should be weighed.[7]
Epidemiology
C2 deficiency has a prevalence of 1 in about 20,000 people in Western countries.[2]
See also
References
- 1 2 Winkelstein, Jerry A. (2004). "Complement Deficiencies". In Crocetti, Michael; Barone, Michael A. (eds.). Oski's Essential Pediatrics (2nd ed.). Philadelphia: Lippincott Williams & Wilkins. p. 670. ISBN 9780781737708. Retrieved 21 September 2016.
- 1 2 3 4 5 6 7 "Complement Deficiencies. What are complement deficiencies?". patient.info. Retrieved 31 December 2017.
- 1 2 3 4 5 "Complement Deficiencies Clinical Presentation: History, Physical, Causes". emedicine.medscape.com. Retrieved 21 September 2016.
- ↑ Winkelstein, Jerry A. (2004). "The Complement System". In Gorbach, Sherwood L.; Bartlett, John G.; Blacklow, Neil R. (eds.). Infectious Diseases. Lippincott Williams & Wilkins. pp. 8–13. ISBN 978-0-7817-3371-7.
- ↑ Sjöholm, A.G.; Jönsson, G.; Braconier, J.H.; Sturfelt, G.; Truedsson, L. (2006). "Complement deficiency and disease: An update". Molecular Immunology. 43 (1–2): 78–85. doi:10.1016/j.molimm.2005.06.025. PMID 16026838. – via ScienceDirect (Subscription may be required or content may be available in libraries.)
- ↑ Moreland, Larry W., ed. (2004). Rheumatology and Immunology Therapy: A to Z Essentials. Berlin: Springer. p. 425. ISBN 9783540206255. Retrieved 30 August 2016.
- 1 2 Complement-Related Disorders at eMedicine
- ↑ Pettigrew, H. David; Teuber, Suzanne S.; Gershwin, M. Eric (September 2009). "Clinical Significance of Complement Deficiencies". Annals of the New York Academy of Sciences. 1173 (1): 108–123. Bibcode:2009NYASA1173..108P. doi:10.1111/j.1749-6632.2009.04633.x. ISSN 1749-6632. PMID 19758139.
- ↑ Aghamohammadi, Asghar; Rezaei, Nima (13 December 2012). Clinical Cases in Primary Immunodeficiency Diseases: A Problem-Solving Approach. Springer Science & Business Media. p. 334. ISBN 978-3-642-31785-9. Retrieved 30 January 2022.
- ↑ "OMIM Entry - # 312060 - PROPERDIN DEFICIENCY, X-LINKED; CFPD". omim.org. Retrieved 21 September 2016.
- ↑ Gower, Richard G; Busse, Paula J; Aygören-Pürsün, Emel; Barakat, Amin J; Caballero, Teresa; Davis-Lorton, Mark; Farkas, Henriette; Hurewitz, David S; Jacobs, Joshua S; Johnston, Douglas T; Lumry, William; Maurer, Marcus (15 February 2011). "Hereditary Angioedema Caused By C1-Esterase Inhibitor Deficiency: A Literature-Based Analysis and Clinical Commentary on Prophylaxis Treatment Strategies". The World Allergy Organization Journal. 4 (Suppl 2): S9–S21. doi:10.1097/1939-4551-4-S2-S9. ISSN 1939-4551. PMC 3666183. PMID 23283143.
- ↑ "Systemic Lupus Erythematosus. Lupus treatment; information | Patient". Patient. Retrieved 21 September 2016.
- ↑ "Membranoproliferative Glomerulonephritis: Background, Pathophysiology, Etiology". Medscape. Retrieved 21 September 2016.
- ↑ Reference, Genetics Home. "C2 gene". Genetics Home Reference. Retrieved 21 September 2016.
- ↑ Reference, Genetics Home. "C3 gene". Genetics Home Reference. Retrieved 21 September 2016.
- ↑ Kolev, Martin V; Ruseva, Marieta M; Harris, Claire L; Morgan, B. Paul; Donev, Rossen M (1 March 2009). "Implication of Complement System and its Regulators in Alzheimer's Disease". Current Neuropharmacology. 7 (1): 1–8. doi:10.2174/157015909787602805. ISSN 1570-159X. PMC 2724661. PMID 19721814.
- ↑ Sekar, Aswin; Bialas, Allison R.; de Rivera, Heather; Davis, Avery; Hammond, Timothy R.; Kamitaki, Nolan; Tooley, Katherine; Presumey, Jessy; Baum, Matthew (11 February 2016). "Schizophrenia risk from complex variation of complement component 4". Nature. 530 (7589): 177–183. Bibcode:2016Natur.530..177.. doi:10.1038/nature16549. ISSN 0028-0836. PMC 4752392. PMID 26814963.
- ↑ Davis, Alvin E.; Mejia, Pedro; Lu, Fengxin (1 October 2008). "Biological activities of C1 inhibitor". Molecular Immunology. 45 (16): 4057–4063. doi:10.1016/j.molimm.2008.06.028. ISSN 0161-5890. PMC 2626406. PMID 18674818.
- ↑ Ram, S.; Lewis, L. A.; Rice, P. A. (7 October 2010). "Infections of People with Complement Deficiencies and Patients Who Have Undergone Splenectomy". Clinical Microbiology Reviews. 23 (4): 740–780. doi:10.1128/CMR.00048-09. ISSN 0893-8512. PMC 2952982. PMID 20930072.
Further reading
- Botto, Marina (1 January 2001). "Links between complement deficiency and apoptosis". Arthritis Research & Therapy. 3 (4): 207–210. doi:10.1186/ar301. ISSN 1478-6362. PMC 128896. PMID 11438036.
- Aghamohammadi, Asghar; Rezaei, Nima (2012). Clinical cases in primary immunodeficiency diseases a problem-solving approach. Berlin: Springer. ISBN 9783642317859. Retrieved 21 September 2016.