| Kearns–Sayre syndrome | |
|---|---|
| Other names | Oculocraniosomatic disorder or Oculocranionsomatic neuromuscular disorder with ragged red fibers |
| Specialty | Ophthalmology |
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia (abbreviated CPEO), a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid (levator palpebrae, orbicularis oculi) and eye (extra-ocular muscles). This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies.[1] In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.
Signs and symptoms
Individuals with KSS present initially in a similar way to those with typical CPEO. Onset is in the first and second decades of life.
The first symptom of this disease is a unilateral ptosis, or difficulty opening the eyelids, that gradually progresses to a bilateral ptosis. As the ptosis worsens, the individual commonly extends their neck, elevating their chin in an attempt to prevent the eyelids from occluding the visual axis. Along with the insidious development of ptosis, eye movements eventually become limited causing a person to rely more on turning the head side to side or up and down to view objects in the peripheral visual field.
Mitochondrial retinopathy

Kearns and Sayre described patients with "pigmentary degeneration" on funduscopy, night vision abnormalities, and some histologic similarities, but also clinical differences, to retinitis pigmentosa[2] Subsequently, the retinal phenotype of KSS was described as retinitis pigmentosa, atypical retinitis pigmentosa, tapetoretinal degeneration, salt-and-pepper retinopathy, and pigmentary retinopathy. As the clinical characterization, however, was not always comprehensive the term "mitochondrial retinopathy" appears most accurate and the diagnosis of RP may have been imprecise.[3] Patients with KSS show widespread granular pigmented alterations in the posterior fundus which correspond to granular patterns on fundus autofluorescence imaging. Associated changes on optical coherence tomography (OCT) include reflectivity changes predominantly at the level of the ellipsoid and interdigitation zone and an increased distance between the ellipsoid band and the retinal pigment epithelium.[3] Night blindness may be seen in patients with KSS. Visual acuity loss is usually mild and only occurs in 40–50% of patients.[4]
Cardiac conduction abnormalities
These most often occur years after the development of ptosis and ophthalmoplegia.[4] Atrioventricular (abbreviated "AV") block is the most common cardiac conduction deficit. This often progresses to a Third-degree atrioventricular block, which is a complete blockage of the electrical conduction from the atrium to the ventricle. Symptoms of heart block include syncope, exercise intolerance, and bradycardia.
Cerebral folate deficiency
Kearns-Sayre patients are consistently found to have cerebral folate deficiency, a syndrome in which 5-MTHF levels are decreased in the cerebrospinal fluid despite being normal in serum.[5] Treatment with folinic acid can in some cases alleviate the associated symptoms and partially correct associated brain abnormalities, especially if started early in the course of illness.[6] The proposed cause of cerebral folate deficiency in the Kearns–Sayre syndrome is the failure of the mechanisms in the choroid plexus that are responsible for passage of folates from the serum to the cerebrospinal fluid.[7]
Cause and prevalence
As characterized in Kearns's original publication in 1965 and in later publications, inconsistent features of KSS that may occur are weakness of facial, pharyngeal, trunk, and extremity muscles, hearing loss, small stature, electroencephalographic changes, cerebellar ataxia and elevated levels of cerebrospinal fluid protein.[8]
Kearns–Sayre syndrome occurs spontaneously in the majority of cases. In some cases it has been shown to be inherited through mitochondrial, autosomal dominant, or autosomal recessive inheritance. There is no predilection for race or sex, and there are no known risk factors. As of 1992 there were only 226 cases reported in published literature.[9] Although NIH and other studies estimate occurrence in the population to be 1–3 and some as high as 9 in 100,000 individuals but a failure to be referred to specialist centres and recognise the disease symptoms is common [9]
Genetics
KSS is the result of deletions in mitochondrial DNA (mtDNA) that cause a particular constellation of medical signs and symptoms. mtDNA is transmitted exclusively from the mother's ovum.[10] Mitochondrial DNA is composed of 37 genes found in the single circular chromosome measuring 16,569 base pairs in length. Among these, 13 genes encode proteins of the electron transport chain (abbreviated "ETC"), 22 encode transfer RNA (tRNA), and two encode the large and small subunits that form ribosomal RNA (rRNA). The 13 proteins involved in the ETC of the mitochondrion are necessary for oxidative phosphorylation. Mutations in these proteins results in impaired energy production by mitochondria. This cellular energy deficit manifests most readily in tissues that rely heavily upon aerobic metabolism such as the brain, skeletal and cardiac muscles, sensory organs, and kidneys. This is one factor involved in the presentation of mitochondrial diseases.
There are other factors involved in the manifestation of a mitochondrial disease besides the size and location of a mutation. Mitochondria replicate during each cell division during gestation and throughout life. Because the mutation in mitochondrial disease most often occurs early in gestation in these diseases, only those mitochondria in the mutated lineage are defective. This results in an uneven distribution of dysfunctional mitochondria within each cell, and among different tissues of the body. This describes the term heteroplasmic which is characteristic of mitochondrial diseases including KSS. The distribution of mutated mtDNA in each cell, tissue, and organ, is dependent on when and where the mutation occurs.[11] This may explain why two patients with an identical mutation in mtDNA can present with entirely different phenotypes and in turn different syndromes. A publication in 1992 by Fischel-Ghodsian et al. identified the same 4,977-bp deletion in mtDNA in two patients presenting with two entirely different diseases. One of the patients had characteristic KSS, while the other patient had a very different disease known as Pearson marrow pancreas syndrome.[12] Complicating the matter, in some cases Pearson's syndrome has been shown to progress into KSS later in life.[13]
More recent studies have concluded that mtDNA duplications may also play a significant role in determining what phenotype is present. Duplications of mtDNA seem to be characteristic of all cases of KSS and Pearson's syndrome, while they are absent in CPEO.[13][14]
Deletions of mtDNA in KSS vary in size (1.3–8kb), as well as position in the mitochondrial genome. The most common deletion is 4.9kb and spans from position 8469 to position 13147 on the genome. This deletion is present in approximately ⅓ of people with KSS[15]
Diagnosis
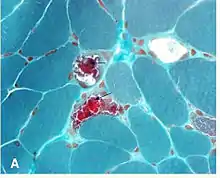
A neuro-ophthalmologist is usually involved in the diagnosis and management of KSS. An individual should be suspected of having KSS based upon clinical exam findings. Suspicion for myopathies should be increased in patients whose ophthalmoplegia does not match a particular set of cranial nerve palsies (oculomotor nerve palsy, fourth nerve palsy, sixth nerve palsy). Initially, imaging studies are often performed to rule out more common pathologies. Diagnosis may be confirmed with muscle biopsy, and may be supplemented with PCR determination of mtDNA mutations.
Biopsy findings
It is not necessary to biopsy an ocular muscle to demonstrate histopathologic abnormalities. Cross-section of muscle fibers stained with Gömöri trichrome stain is viewed using light microscopy. In muscle fibers containing high ratios of the mutated mitochondria, there is a higher concentration of mitochondria. This gives these fibers a darker red color, causing the overall appearance of the biopsy to be described as "ragged red fibers. Abnormalities may also be demonstrated in muscle biopsy samples using other histochemical studies such as mitochondrial enzyme stains, by electron microscopy, biochemical analyses of the muscle tissue (ie electron transport chain enzyme activities), and by analysis of muscle mitochondrial DNA."[16]
Laboratory studies
Blood lactate and pyruvate levels usually are elevated as a result of increased anaerobic metabolism and a decreased ratio of ATP:ADP. CSF analysis shows an elevated protein level, usually >100 mg/dl, as well as an elevated lactate level.[9]
Management
Currently there is no curative treatment for KSS. Because it is a rare condition, there are only case reports of treatments with very little data to support their effectiveness. Several promising discoveries have been reported which may support the discovery of new treatments with further research. Satellite cells are responsible for muscle fiber regeneration. It has been noted that mutant mtDNA is rare or undetectable in satellite cells cultured from patients with KSS. Shoubridge et al. (1997) asked the question whether wildtype mtDNA could be restored to muscle tissue by encouraging muscle regeneration. In the forementioned study, regenerating muscle fibers were sampled at the original biopsy site, and it was found that they were essentially homoplasmic for wildtype mtDNA.[11] Perhaps with future techniques of promoting muscle cell regeneration and satellite cell proliferation, functional status in KSS patients could be greatly improved.
One study described a patient with KSS who had reduced serum levels of coenzyme Q10. Administration of 60–120 mg of coenzyme Q10 for three months resulted in normalization of lactate and pyruvate levels, improvement of previously diagnosed first degree AV block, and improvement of ocular movements.[17]
A screening ECG is recommended in all patients presenting with CPEO. In KSS, implantation of pacemaker is advised following the development of significant conduction disease, even in asymptomatic patients.[18]
Screening for endocrinologic disorders should be performed, including measuring serum glucose levels, thyroid function tests, calcium and magnesium levels, and serum electrolyte levels. Hyperaldosteronism is seen in 3% of KSS patients.[19]
In December 2022 researchers reported a study with modest results in five patients affected by either Pearson syndrome or Kearns–Sayre syndrome.[20][21]
History
The triad of CPEO, bilateral pigmentary retinopathy, and cardiac conduction abnormalities was first described in a case report of two patients in 1958 by Thomas P. Kearns (1922–2011), MD., and George Pomeroy Sayre (1911–1992), MD.[22] A second case was published in 1960 by Jager and co-authors reporting these symptoms in a 13-year-old boy.[23] Previous cases of patients with CPEO dying suddenly had been published, occasionally documented as from a cardiac dysrhythmia. Other cases had noted a peculiar pigmentation of the retina, but none of these publications had documented these three pathologies occurring together as a genetic syndrome.[24] Kearns published a defining case in 1965 describing nine unrelated cases with this triad.[24] In 1988, the first connection was made between KSS and large-scale deletions of muscle mitochondrial DNA (abbreviated mtDNA)[25][26] Since this discovery, numerous deletions in mitochondrial DNA have been linked to the development of KSS.[27][28][29]
References
- ↑ Harvey, J. N.; Barnett, D. (July 1992). "Endocrine dysfunction in Kearns-Sayre syndrome". Clinical Endocrinology. 37 (1): 97–104. doi:10.1111/j.1365-2265.1992.tb02289.x. PMID 1424198. S2CID 24560049.
- ↑ KEARNS TP; SAYRE GP (1958). "Retinitis Pigmentosa, External Ophthalmoplegia, and Complete Heart Block Unusual Syndrome with Histologic Study in One of Two Cases". AMA Archives of Ophthalmology. 60 (2): 280–289. doi:10.1001/archopht.1958.00940080296016. PMID 13558799.
- 1 2 Birtel, Johannes; von Landenberg, Christina; Gliem, Martin; Gliem, Carla; Reimann, Jens; Kunz, Wolfram S.; Herrmann, Philipp; Betz, Christian; Caswell, Richard; Nesbitt, Victoria; Kornblum, Cornelia; Charbel Issa, Peter (2022). "Mitochondrial Retinopathy". Ophthalmology Retina. 6 (1): 65–79. doi:10.1016/j.oret.2021.02.017. PMID 34257060. S2CID 235822866.
- 1 2 Miller, Neil R.; Newman, Nancy J., eds. (2007). Walsh & Hoyt's Clinical Neuro-Ophthalmology: The Essentials. Lippincott Williams & Wilkins. ISBN 9780781763790.
- ↑ Garcia-Cazorla, A.; Quadros, E. V.; Nascimento, A.; Garcia-Silva, M. T.; Briones, P.; Montoya, J.; Ormazabal, A.; Artuch, R.; Sequeira, J. M.; Blau, N.; Arenas, J.; Pineda, M.; Ramaekers, V. T. (2008). "Mitochondrial Diseases Associated with Cerebral Folate Deficiency". Neurology. 70 (16): 1360–1362. doi:10.1212/01.wnl.0000309223.98616.e4. PMID 18413591. S2CID 44622892.
- ↑ Quijada-Fraile, Pilar; o'Callaghan, Mar; Martín-Hernández, Elena; Montero, Raquel; Garcia-Cazorla, Àngels; De Aragón, Ana Martínez; Muchart, Jordi; Málaga, Ignacio; Pardo, Rafael; García-Gonzalez, Pedro; Jou, Cristina; Montoya, Julio; Emperador, Sonia; Ruiz-Pesini, Eduardo; Arenas, Joaquín; Martin, Miguel Angel; Ormazabal, Aida; Pineda, Mercè; García-Silva, María T.; Artuch, Rafael (2014). "Follow-up of folinic acid supplementation for patients with cerebral folate deficiency and Kearns-Sayre syndrome". Orphanet Journal of Rare Diseases. 9: 217. doi:10.1186/s13023-014-0217-2. PMC 4302586. PMID 25539952.
- ↑ Spector, Reynold; Johanson, Conrad E. (2010). "Choroid plexus failure in the Kearns-Sayre syndrome". Cerebrospinal Fluid Research. 7: 14. doi:10.1186/1743-8454-7-14. PMC 2939631. PMID 20731822.
- ↑ Butler, Ian J. (1976-11-01). "Kearns-Sayre Syndrome: A Review of a Multisystem Disorder of Children and Young Adults". Archives of Internal Medicine. 136 (11): 1290–1293. doi:10.1001/archinte.1976.03630110054014. ISSN 0003-9926. PMID 791168.
- 1 2 3 Kearns-Sayre Syndrome at eMedicine
- ↑ Fine, Paule.M. (September 1978). "Mitochondrial Inheritance and Disease". The Lancet. 312 (8091): 659–662. doi:10.1016/S0140-6736(78)92764-2. PMID 80581. S2CID 40192499.
- 1 2 Shoubridge, E.; Johns, T.; Karpati, G. (December 1997). "Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy". Human Molecular Genetics. 6 (13): 2239–2242. doi:10.1093/hmg/6.13.2239. PMID 9361028.
- ↑ Fischel-Ghodsian, Nathan; Bohlman, M Charlotte; Prezant, Toni R.; Graham, John M.; Cederbaum, Stephen D.; Edwards, Matthew J. (June 1992). "Deletion in Blood Mitochondrial DNA in Kearns-Sayre Syndrome". Pediatric Research. 31 (6): 557–560. doi:10.1203/00006450-199206000-00004. PMID 1635816.
- 1 2 Poulton, Joanna; Morten, Karl J.; Weber, Katharina; Brown, Garry K.; Bindoff, Laurence (June 1994). "Are duplications of mitochondrial DNA characteristic of Kearns—Sayre syndrome?". Human Molecular Genetics. 3 (6): 947–951. doi:10.1093/hmg/3.6.947. PMID 7951243.
- ↑ Miller, Neil R.; Newman, Nancy J.; Bioussee, Valerie; Kerrison, John B. (2008). "Ch. 20, adapted from a chapter 22 by Paul N. Hoffman". Walsh and Hoyt's Clinical Neuro-ophthalmology: the essentials. Philadelphia: Lippincott Williams & Wilkins. pp. 432–436. ISBN 9780781763790.
- ↑ PMID 11715003
- ↑ Rubin, Richard M.; Sadun, Alfredo A. (2008). "Ch. 9.17 Ocular Myopathies". In Yanoff, Myron; Duker, Jason (eds.). Ophthalmology (Online Textbook) (3rd ed.). Mosby.
- ↑ Ogasahara, S.; Yorifuji, S.; Nishikawa, Y.; Takahashi, M.; Wada, K.; Hazama, T.; Nakamura, Y.; Hashimoto, S.; Kono, N.; Tarui, S. (1985). "Improvement of abnormal pyruvate metabolism and cardiac conduction defect with coenzyme Ql0 in Kearns-Sayre syndrome". Neurology. 35 (3): 372–377. doi:10.1212/WNL.35.3.372. PMID 3974895. S2CID 27569662.
- ↑ Gregoratos, Gabriel; Abrams, Jonathan; Epstein, Andrew E.; Freedman, Roger A.; Hayes, David L.; Hlatky, Mark A.; Kerber, Richard E.; Naccarelli, Gerald V.; Schoenfeld, Mark H.; Silka, Michael J.; Winters, Stephen L.; Gibbons, Raymond J.; Antman, Elliott M.; Alpert, Joseph S.; Gregoratos, Gabriel; Hiratzka, Loren F.; Faxon, David P.; Jacobs, Alice K.; Fuster, Valentin; Smith, Sidney C. (2002). "ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices: Summary Article". Circulation. 106 (16): 2145–2161. doi:10.1161/01.CIR.0000035996.46455.09. PMID 12379588.
- ↑ Kearns-Sayre Syndrome~diagnosis at eMedicine
- ↑ "Moms' mitochondria may refresh cells in sick kids". www.science.org. Retrieved 2023-01-09.
- ↑ Jacoby, Elad; et al. (21 December 2022). "Mitochondrial augmentation of hematopoietic stem cells in children with single large-scale mitochondrial DNA deletion syndromes". Science Translational Medicine. 4 (676): eabo3724. doi:10.1126/scitranslmed.abo3724. PMID 36542693. S2CID 254998216.
- ↑ Kearns, Thomas P. (1958). "Retinitis Pigmentosa, External Ophthalmoplegia, and Complete Heart Block". AMA Archives of Ophthalmology. 60 (2): 280. doi:10.1001/archopht.1958.00940080296016.
- ↑ Jager, B.V.; Fred, Herbert L.; Butler, Ronald B.; Carnes, William H. (1960). "Occurrence of retinal pigmentation, ophthalmoplegia, ataxia, deafness and heart block". The American Journal of Medicine. 29 (5): 888–893. doi:10.1016/0002-9343(60)90122-4. PMID 13789175.
- 1 2 Kearns, T. P. (1965). "External Ophthalmoplegia, Pigmentary Degeneration of the Retina, and Cardiomyopathy: A Newly Recognized Syndrome". Transactions of the American Ophthalmological Society. 63: 559–625. PMC 1310209. PMID 16693635.
- ↑ Zeviani, M.; Moraes, C. T.; Dimauro, S.; Nakase, H.; Bonilla, E.; Schon, E. A.; Rowland, L. P. (1988). "Deletions of mitochondrial DNA in Kearns-Sayre syndrome". Neurology. 38 (9): 1339–1346. doi:10.1212/wnl.38.9.1339. PMID 3412580. S2CID 30046555.
- ↑ Lestienne, Patrick; Ponsot, Gérard (1988). "Kearns-Sayre Syndrome with Muscle Mitochondrial DNA Deletion". The Lancet. 331 (8590): 885. doi:10.1016/S0140-6736(88)91632-7. PMID 2895391. S2CID 6811844.
- ↑ Zeviani, M.; Moraes, C. T.; Dimauro, S.; Nakase, H.; Bonilla, E.; Schon, E. A.; Rowland, L. P. (1988). "Deletions of mitochondrial DNA in Kearns-Sayre syndrome". Neurology. 38 (9): 1339–1346. doi:10.1212/wnl.38.9.1339. PMID 3412580. S2CID 30046555.
- ↑ Lertrit, P.; Imsumran, A.; Karnkirawattana, P.; Devahasdin, V.; Sangruchi, T.; Atchaneeyasakul, L.; Mungkornkarn, C.; Neungton, N. (1999). "A unique 3.5-kb deletion of the mitochondrial genome in Thai patients with Kearns-Sayre syndrome". Human Genetics. 105 (1–2): 127–131. doi:10.1007/s004390051074. PMID 10480366. Archived from the original on 2000-09-29.
- ↑ Soga, F.; Ueno, S.; Yorifuji, S. (September 1993). "Deletions of mitochondrial DNA in Kearns-Sayre syndrome". Nihon Rinsho. Japanese Journal of Clinical Medicine. 51 (9): 2386–2390. PMID 8411717.
External links
- kearns_sayre at NINDS
- Kearns Sayre syndrome at NIH's Office of Rare Diseases