Transcriptome-wide association study (TWAS) is a genetic methodology that can be used to compare the genetic components of gene expression and the genetic components of a trait to determine if an association is present between the two components.[1][2] TWAS are useful for the identification and prioritization of candidate causal genes in candidate gene analysis following genome-wide association studies.[3] TWAS looks at the RNA products of a specific tissue and gives researchers the abilities to look at the genes being expressed as well as gene expression levels, which varies by tissue type. TWAS are valuable and flexible bioinformatics tools that looks at the associations between the expressions of genes and complex traits and diseases.[4] By looking at the association between gene expression and the trait expressed, genetic regulatory mechanisms can be investigated for the role that they play in the development of specific traits and diseases.
Transcriptome Analysis
A transcriptome is the sum of all RNA transcripts that are present in a given cell, tissue, or organ within an organism. Transcriptomes include both mRNA, which functions as an intermediate to the central dogma; as well as noncoding RNAs that may play other roles in protein synthesis.[5] In the central dogma, it describes how DNA is able to make proteins through transcription and translation. RNAs are present in a cell in varied concentrations, and play various roles outside of the central dogma and are able to be identified based on length and function. It is through functional elements that the transcriptional and translational activities of genes is able to be regulated.[4] Transcriptome analysis is beneficial for obtaining information about all RNAs present and can provide valuable insight into the genetic mechanisms that are tissue specific.[6] The transcriptome was first investigated in the 1990s in an experiment performed to identify a partial transcriptome of the human brain. Researchers were able to identify 609 mRNA sequences.[5] Since then, many advances in Next Generation Sequencing methods have been made.[6] Transcriptomes are now able to be routinely developed due to advances in these methods and new technologies such as microarrays and RNA-Seq. Both methods require computed imaging as well as high reads and statistical analysis.[5] By obtaining information about gene expression through mRNAs, many applications have been discovered. Transcriptome analysis has proven to be beneficial in identifying disease processes as well as regulatory elements in disease progressions, has aided drug development through identification of disease processes, offers insight into therapeutic strategies, and has improved identification of genes that are able to respond to both biotic and abiotic environmental factors as well as how environmental conditions play a role in gene expression.[5]
Methods
A genome-wide association study, or GWAS, is a genetic tool that uses single nucleotide polymorphisms, or SNPs, to identify if a trait or disease is linked to a specific genetic variant. By observing if frequencies of a specific variant are more commonly associated, or higher than expected, with the given trait; an association is developed between the trait and the variant. However, many of these associations can be developed throughout an individual due to linkage disequilibrium and the large size of the genome. Although GWAS provide valuable insight into identifying markers throughout the genome, a large portion of the SNPs are present in non-coding mRNA regions, and many have unknown functions that are difficult to determine through standard methods, as no product is manufactured by these regions of the genome.[4]
Transcription-wide association studies are able to take the information from GWAS results and are able to utilize these results as reference data, and can then help to identify and prioritize genes.[4] In order to perform this analysis, a reference panel for gene expression should be obtained, such as an expression quantitative trait loci (eQTL), which helps to show gene expression regulation.[4] Using the reference panel, a predictive model can be generated to impute the expression variation of genes. Imputation is the process by which you can predict the expression levels of genes in other organisms through the variation that exists in their genome based on a reference panel. By predicting the levels of gene expression within a tissue, other variables such as environment and epigenetic effects are eliminated as this prediction is solely based on the variant present and the expected level of gene expression. However, this can lead to inaccuracies with gene expression predictions as both the environment and epigenetic modifications are able to alter the level of gene expression.[7] Cis-genetic components are the primary focus of the eQTLs as these are elements that are within 1 Megabase of a gene.[2] The predictive model can then be applied to the GWAS samples to predict gene expression of the significant SNPs from that study. After expression levels are imputated, gene-trait association tests are performed by associating levels of predicted expression and genotypes with the phenotypes of the individual.[8] Essentially, a TWAS is able to take GWAS results and are able to predict the effects of each variant on expression levels of genes that are associated with the trait, and it is able to do this for every loci that is associated with the trait throughout the genome.[7]
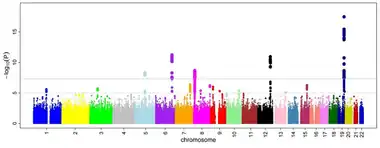
Results are able to be interpreted on a Manhattan plot, similarly to GWAS results. Any loci that are considered to have statistically significant results will have a higher P value, and this indicates that the loci is likely associated with the trait or disease being investigated. Any statistically significant P-values have a higher log P-value and show above the Bonferroni correction line. The Manhattan plot is named as such as the statistically significant genes appear to show up as "skyscrapers" on the plot, and when there are many genes that are associated with the trait, the plot resembles the Manhattan skyline. Although the Manhattan plot image is for a GWAS study, TWAS results are shown the same way. Statistically significant loci are genes that have significantly associated SNPs whose expression correlates with the trait or disease of interest.
PrediXcan[1] and FUSION[2] are both TWAS software that have been utilized in genetic studies to investigate the gene-trait associations. PrediXcan is a well-developed TWAS software that has the ability to estimate genetically regulated expression and determine associations with the phenotype being investigated. It uses a penalized regression model to give weight to levels of observed gene expression and cis-SNPs derived from the reference dataset.[8] The software then uses individual genotype data to perform gene-trait association tests. FUSION is another TWAS software that utilizes a different statistical analysis to create the association tests. In this model, imputation methods from the predictive model are calculated based on a Bayesian sparse linear mixed model.[8] The advantage of this software is that it is able to perform association tests on individual genotype data, but this software can also take information from large scale data sets using imputation.
Advantages
The advantages of this methodology are through the insight it gives researchers into the function of genes and the association between gene functions and gene expression. TWAS has the potential to take results from GWAS and extend the results to aid in the understanding of disease mechanisms.[4] Additionally, as this method uses loci that were previously identified by GWAS analysis, there is a lower testing burden associated with a TWAS as less sites are analyzed. By lowering the number of loci being analyzed, this allows more in-depth analysis of the sites analyzed and can give further insight to the functions and associations of the significant loci.
TWAS also have the advantage of reducing the effects of confounding factors. When building a predictive model, it only looks at genetic expression, not total expression. Total expression includes factors like the environment and epigenetic modifications to levels of expression, and are not accounted for in the predictive model. By not accounting for these factors, it can reduce the accuracy of predicted levels of gene expression; however, it also reduces the effects of confounding variables in the results.[2]
Another advantage of TWAS is that the results are tissue specific. The level of gene expression differs by the tissue that the genes are in, as each tissue has specific splicing patterns and patterns of regulation. By having tissue specific results, this furthers the information that can be derived through these studies as results have the ability to show how gene regulation differs by tissue types as well as how functions are regulated and if there are common regulatory mechanisms between tissues or if regulatory mechanisms have different functions in different tissues. TWAS cross tissue methods also have the possibility to identify potential causal genes for diseases and traits on a larger scale, however, single tissue methods have the ability to determine associations on a case specific basis.[8]
Limitations
Many of the disadvantages of TWAS are implications of the prediction capabilities of the model used to predict gene expression levels based on genotypes. One disadvantage of TWAS is that it mainly looks at cis-genetic components for imputation and for in most studies, does not identify any trans-genetic component variants. This acts as a disadvantage for TWAS as trans-genetic component variants are any regulatory mechanisms that are outside of a 1 Megabase range of the gene, and even though they are a significant distance away from the gene of interest, many regulatory mechanisms have the potential to act long range and can still impact expression. By not taking these components into account, it lowers the accuracy of predicted genetic expression levels and can cause deviation between expected and observed expression levels. As mentioned above, another disadvantage of these studies are that environmental and epigenetic mechanisms for regulation of gene expression is not taken into account with the predictive model for gene expression, which also has the potential to lead to inaccuracies with the predicted gene expression levels and observed expression levels. Another challenge for TWAS is that it can be hard to predict accurate gene expression levels when genes have low heritability levels. eQTLs rely on a level of heritability, and when low heritability is observed, it can affect the observance of false positives and can negatively impact the prediction capabilities of the model used for TWAS.[4]
Additionally, another challenge for TWAS, very similar to GWAS results, is that these studies can only demonstrate associations from results. Even though a statistically significant association can be seen between the gene or loci of interest and the trait or disease, no causal relationship can be derived. In order to establish a causal relationship, further studies utilizing a reverse genetics approach for knock-outs of genes or site-directed mutagenesis would need to be performed to identify causal relationships.
Another issue with TWAS results are the implications of tissue bias and coregulation. Due to the specificity of genetic regulation mechanisms within each tissue, many experiments would need to be performed to determine the tissue specific nature of each loci association and how these associations differ between tissue types.[3] Co-regulation results from a regulatory mechanism controlling the expression of more than one loci at a time. By controlling more than one loci, associations may be drawn between the loci of interest along with other genes or loci that are solely controlled by the same mechanism and may not have any association with the trait or disease of interest, leading to false positive results.[3]
Applications
Schizophrenia
A TWAS study was performed following a GWAS investigating loci associated with schizophrenia. From the GWAS results, over 100 risk loci were located. A TWAS was then used to identify 157 significant loci using expression data, and 35 of the identified loci from the TWAS did not align with the GWAS loci. Results were then further narrowed using regulatory target investigations. 42 of these genes were found to have a statistically significant association with chromatin phenotypes, which is a regulatory mechanism that could further be investigated. MAPK3 was one association that was observed to have a large impact on neurodevelopmental phenotypes, and was further prioritized as a candidate causal gene.[9]
Breast Cancer
In 2018, a TWAS was used to identify candidate causal genes for breast cancer. Data was collected from The Cancer Genome Atlas to establish genetic models as well as 229,000 women of European ancestry. In this study, 8,597 genes were evaluated. Through GWAS studies, around 170 loci were associated with at least one variant of breast cancer. In this study, 179 genes were found to have an association with a variant of breast cancer. Of the 179 genes with associations, 48 were identified to be statistically significant using a Bonferroni-correction threshold (as seen on the Manhattan plot above).[10] 14 of these had never been reported to be associated with a risk of breast cancer previously. The other 34 genes at known risk loci had 23 that do not have any associated risk SNPs.[10] Using gene knock-downs, 13 genes with high predicted levels of expression were found to be associated with an increased risk. When knocked-down, studies showed that 11 of the genes investigated had an effect in a cell line of breast cancer, especially in 184A1 normal breast cells.[10] These genes include the following: PIDD1, NRBF2, and ABHD8.[10] All of the genes identified in the study, both up- and down-regulated had relatively high cis-heritability.
Parkinson's Disease
A TWAS study was completed in 2021 that utilized the most recent Parkinson's Disease (PD) GWAS that utilized 480.000 individuals. From those results, 18 genes were found to have a statistically significant association with PD. The most significant of these was LRRC37A2, which was found to be associated in all 13 brain tissues.[11]
TWAS Atlas
TWAS Atlas is a site that has been curated to integrate the findings of many TWAS studies. This atlas exists virtually and is accessible to the public. Results and findings that are published in the TWAS Atlas are able to be integrated and combined to aid future studies and the understanding of genetic regulation mechanisms. Results are presented in a visual manner to improve the integration of results. Currently, 401,226 TWAS associations have been published from 200 publications, spanning 257 traits and 22,247 genes as of April 25, 2022.[12]
References
- 1 2 Gamazon, Eric R.; Wheeler, Heather E.; Shah, Kaanan P.; Mozaffari, Sahar V.; Aquino-Michaels, Keston; Carroll, Robert J.; Eyler, Anne E. (August 2015). "A gene-based association method for mapping traits using reference transcriptome data". Nature Genetics. 47 (9): 1091–1098. doi:10.1038/ng.3367. ISSN 1546-1718. PMC 4552594. PMID 26258848.
- 1 2 3 4 Gusev, Alexander; Ko, Arthur; Shi, Huwenbo; Bhatia, Gaurav; Chung, Wonil; Penninx, Brenda W. J. H.; Jansen, Rick; de Geus, Eco J. C.; Boomsma, Dorret I.; Wright, Fred A.; Sullivan, Patrick F.; Nikkola, Elina; Alvarez, Marcus; Civelek, Mete; Lusis, Aldons J. (March 2016). "Integrative approaches for large-scale transcriptome-wide association studies". Nature Genetics. 48 (3): 245–252. doi:10.1038/ng.3506. ISSN 1546-1718. PMC 4767558. PMID 26854917.
- 1 2 3 Wainberg, Michael; Sinnott-Armstrong, Nasa; Mancuso, Nicholas; Barbeira, Alvaro N.; Knowles, David A.; Golan, David; Ermel, Raili; Ruusalepp, Arno; Quertermous, Thomas; Hao, Ke; Björkegren, Johan L. M.; Im, Hae Kyung; Pasaniuc, Bogdan; Rivas, Manuel A.; Kundaje, Anshul (April 2019). "Opportunities and challenges for transcriptome-wide association studies". Nature Genetics. 51 (4): 592–599. doi:10.1038/s41588-019-0385-z. ISSN 1546-1718. PMC 6777347. PMID 30926968.
- 1 2 3 4 5 6 7 Li, Binglan; Ritchie, Marylyn D. (2021-09-30). "From GWAS to Gene: Transcriptome-Wide Association Studies and Other Methods to Functionally Understand GWAS Discoveries". Frontiers in Genetics. 12: 713230. doi:10.3389/fgene.2021.713230. ISSN 1664-8021. PMC 8515949. PMID 34659337.
- 1 2 3 4 Lowe, Rohan; Shirley, Neil; Bleackley, Mark; Dolan, Stephen; Shafee, Thomas (2017-05-18). "Transcriptomics technologies". PLOS Computational Biology. 13 (5): e1005457. Bibcode:2017PLSCB..13E5457L. doi:10.1371/journal.pcbi.1005457. ISSN 1553-734X. PMC 5436640. PMID 28545146.
- 1 2 Jiang, Zhihua; Zhou, Xiang; Li, Rui; Michal, Jennifer J.; Zhang, Shuwen; Dodson, Michael V.; Zhang, Zhiwu; Harland, Richard M. (September 2015). "Whole transcriptome analysis with sequencing: methods, challenges and potential solutions". Cellular and Molecular Life Sciences. 72 (18): 3425–3439. doi:10.1007/s00018-015-1934-y. ISSN 1420-9071. PMC 6233721. PMID 26018601.
- 1 2 Cao, Chen; Ding, Bowei; Li, Qing; Kwok, Devin; Wu, Jingjing; Long, Quan (February 2021). "Power analysis of transcriptome-wide association study: Implications for practical protocol choice". PLOS Genetics. 17 (2): e1009405. doi:10.1371/journal.pgen.1009405. ISSN 1553-7404. PMC 7946362. PMID 33635859.
- 1 2 3 4 Xie, Yuhan; Shan, Nayang; Zhao, Hongyu; Hou, Lin (2021-06-15). "Transcriptome wide association studies: general framework and methods". Quantitative Biology. 9 (2): 141–150. doi:10.15302/J-QB-020-0228. ISSN 2095-4689. S2CID 234134491.
- ↑ Gusev, Alexander; Mancuso, Nicholas; Won, Hyejung; Kousi, Maria; Finucane, Hilary K.; Reshef, Yakir; Song, Lingyun; Safi, Alexias; McCarroll, Steven; Neale, Benjamin M.; Ophoff, Roel A.; O’Donovan, Michael C.; Crawford, Gregory E.; Geschwind, Daniel H.; Katsanis, Nicholas (April 2018). "Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights". Nature Genetics. 50 (4): 538–548. doi:10.1038/s41588-018-0092-1. ISSN 1546-1718. PMC 5942893. PMID 29632383.
- 1 2 3 4 Wu, Lang; Shi, Wei; Long, Jirong; Guo, Xingyi; Michailidou, Kyriaki; Beesley, Jonathan; Bolla, Manjeet K.; Shu, Xiao-Ou; Lu, Yingchang; Cai, Qiuyin; Al-Ejeh, Fares; Rozali, Esdy; Wang, Qin; Dennis, Joe; Li, Bingshan (July 2018). "A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer". Nature Genetics. 50 (7): 968–978. doi:10.1038/s41588-018-0132-x. ISSN 1546-1718. PMC 6314198. PMID 29915430.
- ↑ Yao, Shi; Zhang, Xi; Zou, Shu-Cheng; Zhu, Yong; Li, Bo; Kuang, Wei-Ping; Guo, Yan; Li, Xiao-Song; Li, Liang; Wang, Xiao-Ye (2021-09-09). "A transcriptome-wide association study identifies susceptibility genes for Parkinson's disease". npj Parkinson's Disease. 7 (1): 79. doi:10.1038/s41531-021-00221-7. ISSN 2373-8057. PMC 8429416. PMID 34504106.
- ↑ "TWAS Atlas: a curated knowledgebase of transcriptome-wide association studies". Nucleic Acids Res. [PMID 36243959].