1-Phosphaallenes is are allenes in which the first carbon atom is replaced by phosphorus, resulting in the structure: -P=C=C<.
Introduction
The first example of a stable heteroalkene E=C or E=E' (E, E' = P) containing a heavy group 15 element was reported in 1982 by Yoshifuji.[1][2] After the realization of heavier heteroalkenes, the field of organometallic chemistry began exploring the idea of heteroallenes E=C=E' and E=C=C=E', in which one or more carbon atom of an allene is substituted by a heavier atom. The first of these heteroallenes containing a heavier group 15 element to be reported was synthesized in 1984 by Yoshifuji.[3] 1-phosphaallene, also referred to as ethenylidenephosphine or λ3-phosphaallene, was first synthesized using the 2,4,6-tri-t-butylphenyl (supermesityl, or Mes*) moiety as a sterically protecting group.[4][5] The most widely known example of 1-phosphaalenes is the Mes* substituted 3H-phosphallene, where a hydrogen is bonded to the terminal carbon atom, which was synthesized by Märkl and Reitinger in 1988.[6][7] Some more recent advances made in regards to 1-phosphaallenes include the development of simpler synthetic routes and the discovery of synthetic pathways using phosphaallenes to create molecules that are typically only synthesized through complicated methods.
Synthesis
Multiple synthetic routes to 1-phosphaallenes have been reported. The most common are Phospha-Wittig-Horner reactions, rearrangement of alkylynlphosphines with a base present, dehydrochlorination of a chlorovinylphospine, and the reaction of lithium with a C,C-dichlorophosphirane.[3][4] More recent synthetic advances include the dehydrobromination of phosphaalkenes the hydroalumination of alkynylphosphines.[8][9]
Phospha-Wittig-Horner reaction
The Phospha-Wittig-Horner reaction, first reported by Mathey et al. in 1992, was initially developed as a synthetic route to convert aldehydes and ketones into C-mono and C,C-disubstituted phosphaalkenes, respectively.[10][11] This method was then adapted to rapidly synthesize phosphaallenes by reacting phosphaketenes with an organolithium base, such as lithium diisopropylamide or t-BuOLi.[4][10][12]
Rearrangement of alkynylphosphines

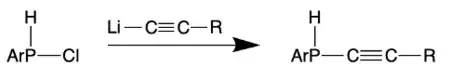
Like the transition from an alkyne to an allene in organic chemistry, phosphaallenes can be synthesized from alkynylphosphines by adding a strong base such as n-butyllithium (n-BuLi). These complexes are typically useful due to the broad range of stable products that can be obtained from this synthetic route.[3][4][13]
Multiple stable phosphaallenes have been synthesized from LiC≡CR and ArP(H)Cl (R = Ph, t-Bu, Me, CH2OSiMe3). The basicity of the lithium complex is essential to the success of these reactions.[4]
Dehydrochlorination of a chlorovinylphosphine
The dehydrochlorination of 1-chlorovinylphosphine by DBU (1,8-diazabicyclo-[5.4.0]undec-7-ene) yields a variety of results, depending on the structure of the initial chlorovinylphosphine. Many of these reactions require temperatures between -90 °C and 0 °C to ensure a stable product is formed. As many of these reactions are warmed, oligomerization of the phosphaallene occurs. Other reactions following this synthetic route occur with gas phase reagents at 250 °C.[4][14]
Reaction of Lithium with a C,C-dichlorophosphirane
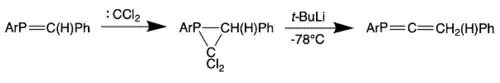
In this synthetic route, a dichlorocarbene is first reacted with a phosphaethylene. The addition of t-BuLi to the resulting 2,2-dichlorophosphirane yields the phosphaallene. This route includes an insertion of carbon into the P=C bond of a phosphaalkene.[4][15]
Dehydrobromination of phosphaalkenes
(Z)-2,5-dibromo-1-phosphapent-1-ene with the sterically cumbersome Mes* moiety was reacted with t-BuOK to yield the cyclopropylidenephosphaethene product. Treatment of the (Z)-2,5-dibromo-1-phosphapent-1-ene reagent with a weaker base, EtONa yields a non-cyclic isomer that can undergo cyclization in the presence of t-BuOK.[8]

Hydroalumination of alkynylphosphines
In 2019, a new synthetic route for the synthesis of phosphaallenes was reported by Klöcker and coworkers. The hydroalumination of dialkynylphosphines with different alkylaluminium hydrides resulted in the elimination of dialkylaluminium alkynides and the desired phosphaallenes. Many of the phosphaallenes Klöcker and coworkers synthesized spontaneously decomposed into less-desired products, but the newly reported synthetic method did allow for the isolation of some highly reactive compounds that had not been isolated previously.[9]

Reactivity
In 2022, the most notable aspect of 1-phosphaallenes is their reactivity. Phosphaallenes contain three reactive centers: the P=C bond, the C=C bond, and the phosphorus atom. The P=C bond and the C=C bond are thought to be electronically independent of one another. This gives rise to a wide variety of reactivity, which is similar to that of phosphaalkenes.
Dimerization
Without a large steric hindrance, most phosphaallenes quickly dimerize in a [2+2]- or [2+3]-cycloaddition to yield four- or five-membered rings.[16] Thus, in order to prevent dimerization, a bulky moiety such as Mes* is often used. Dimerization frequently occurs in a head-to-tail manner, exhibiting coupling between either the two C=C bonds or the two P=C bonds. The regioselectivity of these dimerization reactions depends on the substituents on the C in the 3 position.[3]

Reaction with alkoxide or lithium compounds
Nucleophilic alkoxide as well as n-BuLi will add to the P=C bond of a phosphaallene. As seen in the reaction scheme, the central C atom is the preferred location for proton attack due to the partial negative charge it possesses.[4][9][13][17][18][19]

Reactions involving carbenes
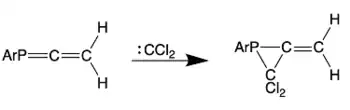
The reaction of phosphaallenes with dichlorocarbenes results in a [2+1]-cycloaddition to the P=C double bond, which results in stable heterocyclic compounds.[4][20]
Phosphaallenes have also been investigated as reagents to form carbenes. Reacting the phosphaallene H2P=C=PH with acetylene yields a carbene via [3 + 2]-cycloaddition.[21]

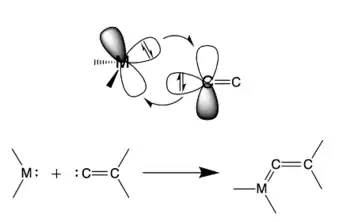
Reaction with transition metal complexes
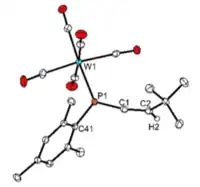
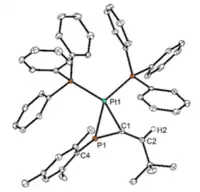
Phosphaallenes can coordinate in an ɳ1 or ɳ2 fashion depending on the metal and experimental conditions. Recent studies into the coordination behavior of the 3H-phosphaallene with tungsten (W) show that both stable and unstable phosphaallenes can bond to W through and ɳ1-coordination mode via a P-W bond.[9][13] Due to the stabilizing effect of coordination, reacting transient phosphaallenes with W has become a common method to create stable phosphaallenes that can then be characterized. These studies have also demonstrated that 3H-phosphaallenes exhibit ɳ2-coordination to Pt via side-on overlap between Pt and the P=C double bond resulting in a three-membered PtPC heterocycle.[9][13][22]
Decomposition
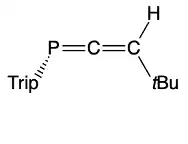
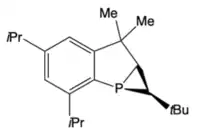
Decomposition of the 3H-phosphaallene occurs at room-temperature to produce several products via P=C (head-to-tail) or C=C dimerization.[15] One decomposition pathway results in an unusual tricyclic phosphine which can be selectively formed by irradiating a solution of the phosphaallene, iBu2AlH, and TripP(C≡CtBu)2 in n-hexanes for 60h with a mercury vapor lamp. Under these conditions, the tricyclic phosphine was isolated in high yield and a purity of >95%.[16]
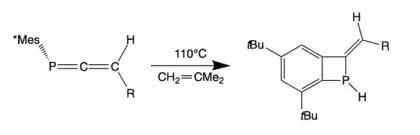
Formation of bicyclic hydrophosphine
Recently, two novel 1-benzodihydrophosphetes were synthesized by heating 3H-phosphaallene Mes*P=C=C(H)R (R = tBu, Ad) in toluene at 110 °C for 16-20h. The resulting 1- benzodihydrophosphetes were not stable unless coordinated to tungsten.[16] The phosphorus-bound H of the product can be used in catalyst-free hydrophosphination reactions and in the synthesis of bulky phosphine ligands.[19]
Hydroboration of the C=C Bond
Hydroboration is used as a method to reduce unsaturated compounds, and has been used to reduce P-C double or triple bonds in phosphaalkenes and phosphaalkynes. Hydroboration of the 3H-phosphaallene Mes*-P=C=C(H)-tBu with 1:1 molar ratio of phosphaallene to H3B(SMe2) yielded a di(phosphaalkenyl)borane, and the reaction in 2:1 molar ratio of phosphaallene to H3B(SMe2) yielded the desired product with a B atom bound to two P=C double bonds. The hydroboration of the C=C bond was preferred over the hydroboration of the P=C bond.[19]

Properties
P=C=C bonding
Phosphaallenes are typically colorless or pale yellow in color, and are often crystalline in structure; however, few crystal structures have been reported for this class of compounds.[4] One compound, ArP=C=CPh2 (Ar =Mes*) was synthesized and characterized by single crystal X-ray diffraction. This compound showed a relatively short P=C double bond length of 1.625(4)Å and a short C=C bond length of 1.1327(5)Å, which closely resembles the bond length in an allene.[23]
The crystal structure also showed a bent geometry with the P=C=C bond angle at 168.0(3)°.[23] This bent geometry is thought to be a result of both steric and electronic effects. One possible explanation for the observed bent geometry is the bonding model postulated by Lappert et al. which described the bonding between P and the C as singlet states that act in a double π-donor-acceptor fashion.[24][25][26][27]
The crystal structure of another 1-phosphaallene RP=C=CHR' (R= Mes*, R' = tBu) displays the same bent geometry with the P=C=C bond angle being reported as 170.7°. Further, this compound also contains a short P=C bond length (1.634Å) and a short C=C bond length (1.136Å).[9]

Electronic structure
The electronic structure of 1-phosphaallene is nearly identical to that of phosphaalkene. The HOMO in both molecules is the π-orbital of the C=P bond, which is primarily P in character. The LUMO in both structures, is also heavily localized over the C=P bond. However, the dipole moment in phosphaallenes is larger than that of phosphaalkenes.[4]

Vibrational frequencies
In compounds with the antisymmetric form X=C=Y, IR stretching bands are observed in the 1600–2300 cm−1 region. However, the IR spectra of 1-phosphaallenes do not contain bands in this region, which suggests that the phosphaallene P=C=C is relatively symmetric. In the phosphaallene H2C=C=PH, stretching frequencies are observed at 1788 and 789 cm−1, which are attributed to the C=C bond and the C=P bond, respectively. In similar compounds containing a C=C=N skeleton, there is a very strong band present within the 1600 – 2300 cm−1 region. The presence of two stretching frequencies rather than one strong frequency in the phosphaallene supports that the C=C and C=P bonds are nearly independent of one another electronically.[4]
References
- ↑ Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. (July 1981). "Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene: isolation of a true phosphobenzene". Journal of the American Chemical Society. 103 (15): 4587–4589. doi:10.1021/ja00405a054.
- ↑ Yoshifuji, Masaaki; Shima, Ichiro; Inamoto, Naoki; Hirotsu, Ken; Higuchi, Taiichi (November 1982). "Additions and Corrections – Synthesis and Structure of Bis(2,4,6-tri-tert-butylphenyl)diphosphene: Isolation of a True "Phosphobenzene"". Journal of the American Chemical Society. 104 (22): 6167. doi:10.1021/ja00386a605.
- 1 2 3 4 Escudié, Jean; Ranaivonjatovo, Henri (1 March 2007). "Group 14 and 15 Heteroallenes ECC and ECE'". Organometallics. 26 (7): 1542–1559. doi:10.1021/om0610086.
- 1 2 3 4 5 6 7 8 9 10 11 12 Escudié, Jean; Ranaivonjatovo, Henri; Rigon, Leslie (1 October 2000). "Heavy Allenes and Cumulenes ECE' and ECCE' (E = P, As, Si, Ge, Sn; E' = C, N, P, As, O, S)". Chemical Reviews. 100 (10): 3639–3696. doi:10.1021/cr990013z. PMID 11749324.
- ↑ Yoshifuji, Masaaki; Toyota, Kozo; Inamoto, Naoki (1984). "Isolation and characterisation of the first stable diphospha-allene: P,P′-bis(2,4,6-tri-t-butylphenyl)-1,3-diphospha-allene". J. Chem. Soc., Chem. Commun. (11): 689–690. doi:10.1039/C39840000689.
- ↑ Klöcker, Hans; Layh, Marcus; Hepp, Alexander; Uhl, Werner (2016). "Hydroalumination of alkynyl-aminophosphines as a promising tool for the synthesis of unusual phosphines: P–N bond activation, a transient phosphaallene, a zwitterionic AlP 2 C 2 heterocycle and a masked Al/P-based frustrated Lewis pair". Dalton Transactions. 45 (5): 2031–2043. doi:10.1039/C5DT02825F. PMID 26391528.
- ↑ Märkl, G.; Reitinger, S. (January 1988). "3H-phosphaallen – alkinyl-1H-phosphan – tautomere". Tetrahedron Letters. 29 (4): 463–466. doi:10.1016/S0040-4039(00)80122-7.
- 1 2 Ito, Shigekazu; Kimura, Shigeo; Yoshifuji, Masaaki (1 April 2003). "Preparation, Structure, and Reaction of a Sterically Encumbered 1-Phosphaallene Containing a Cyclopropylidene Moiety". Organic Letters. 5 (7): 1111–1114. doi:10.1021/ol0341750. PMID 12659586.
- 1 2 3 4 5 6 7 Klöcker, Hans; Tendyck, Jonas C.; Keweloh, Lukas; Hepp, Alexander; Uhl, Werner (27 March 2019). "3H-Phosphaallenes Revisited: Facile Synthesis by Hydroalumination of Alkynylphosphines and β-Elimination, Stability and Trapping of Transient Species by Coordination to Transition Metal Atoms". Chemistry – A European Journal. 25 (18): 4793–4807. doi:10.1002/chem.201806334. PMID 30681211. S2CID 59252113.
- 1 2 Arkhypchuk, Anna I.; Svyaschenko, Yurii V.; Orthaber, Andreas; Ott, Sascha (17 June 2013). "Mechanism of the Phospha-Wittig-Horner Reaction". Angewandte Chemie International Edition. 52 (25): 6484–6487. doi:10.1002/anie.201301469. PMC 3738942. PMID 23653134.
- ↑ De Vaumas, Rene; Marinetti, Angela; Ricard, Louis; Mathey, Francois (January 1992). "Use of prochiral phosphaalkene complexes in the synthesis of optically active phosphines". Journal of the American Chemical Society. 114 (1): 261–266. doi:10.1021/ja00027a034.
- ↑ Appel, Rolf; Fölling, Peter; Josten, Bernhard; Siray, Mustafa; Winkhaus, Volker; Knoch, Falk (August 1984). "Phosphaallenes". Angewandte Chemie International Edition in English. 23 (8): 619–620. doi:10.1002/anie.198406191.
- 1 2 3 4 Ito, Shigekazu; Toyota, Kozo; Yoshifuji, Masaaki (2000). "Reactions of a Sterically Protected 2,3-Dichloro-1,4-Diphospha-1,3-Butadiene with Some Alkyl and Hydrido Lithium Reagents". Heteroatom Chemistry. 11 (3): 171–176. doi:10.1002/(SICI)1098-1071(2000)11:3<171::AID-HC1>3.0.CO;2-6.
- ↑ Guillemin, Jean-Claude; Janati, Tajdine; Denis, Jean-Marc (1992). "Lewis base-induced rearrangement of primary ethyn-1-ylphosphines, a new and efficient route to phosphaalkynes". Journal of the Chemical Society, Chemical Communications (5): 415. doi:10.1039/C39920000415.
- 1 2 Yoshifuji, Masaaki; Yoshimura, Hideki; Toyota, Kozo (May 1990). "Reaction of Dichlorocarbene with Phosphaethylenes. Preparation of Phosphaallene from Phosphaethylene via Dichlorophosphirane". Chemistry Letters. 19 (5): 827–830. doi:10.1246/cl.1990.827.
- 1 2 3 Tendyck, Jonas C.; Klöcker, Hans; Schürmann, Lina; Würthwein, Ernst-Ulrich; Hepp, Alexander; Layh, Marcus; Uhl, Werner (20 November 2020). "Aspects of Phosphaallene Chemistry: Heat-Induced Formation of 1,2-Dihydrophosphetes by Intramolecular Nucleophilic Aromatic Substitution and Photochemical Generation of Tricyclic Phosphiranes". The Journal of Organic Chemistry. 85 (22): 14315–14332. doi:10.1021/acs.joc.9b03056. PMID 32022561. S2CID 211036465.
- ↑ Hafner, Michael; Wegemann, Thomas; Regitz, Manfred (1993). "Organophosphorus Compounds; 72. 1 The Tri- tert -butylcyclopropenyl Group as a Steric Protecting Function for 1-Phosphaallenes". Synthesis. 1993 (12): 1247–1252. doi:10.1055/s-1993-26036.
- ↑ Nguyen, Minh Tho; Hegarty, Anthony F. (1985). "Structure and properties of 1-phospha-allene (H2C=C=PH). α-Carbon versus phosphorus protonation?". J. Chem. Soc., Perkin Trans. 2 (12): 1999–2004. doi:10.1039/P29850001999.
- 1 2 3 Tendyck, Jonas C.; Hepp, Alexander; Würthwein, Ernst-Ulrich; Uhl, Werner (4 December 2020). "New Reactivity Patterns in 3H-Phosphaallene Chemistry [Aryl-P=C=C(H)- t Bu]: Hydroboration of the C=C Bond, Deprotonation and Trimerisation". Chemistry – A European Journal. 26 (68): 15977–15988. doi:10.1002/chem.202002506. PMC 7756809. PMID 32618025.
- ↑ Yoshifuji, Masaaki; Shibata, Manabu; Toyota, Kozo; Miyahara, Ikuko; Hirotsu, Ken (June 1994). "Reaction of dichlorocarbene with 1-(2,4,6-tri-t-butylphenyl)-1-phosphaallenes". Heteroatom Chemistry. 5 (3): 195–200. doi:10.1002/hc.520050302.
- ↑ Fernández, Israel; Cossío, Fernando P.; Bickelhaupt, F. Matthias (1 April 2011). "Aromaticity and Activation Strain Analysis of [3 + 2] Cycloaddition Reactions between Group 14 Heteroallenes and Triple Bonds". The Journal of Organic Chemistry. 76 (7): 2310–2314. doi:10.1021/jo102572x. PMID 21388217.
- ↑ Nixon, John F. (November 1988). "Coordination chemistry of compounds containing phosphorus-carbon multiple bonds". Chemical Reviews. 88 (7): 1327–1362. doi:10.1021/cr00089a015.
- 1 2 Yoshifuji, Masaaki; Toyota, Kozo; Inamoto, Naoki; Hirotsu, Ken; Higuchi, Taiichi; Nagase, Shigeru (December 1985). "Structures of Phosphaethylenes and a 1-Phosphaallene Containing Phosphorus in Lower Coordination State". Phosphorus and Sulfur and the Related Elements. 25 (3): 237–243. doi:10.1080/03086648508072740.
- ↑ Eichler, Barrett; West, Robert (2000). "Chemistry of group 14 heteroallenes". Advances in Organometallic Chemistry. 46: 1–46. doi:10.1016/S0065-3055(00)46001-6. ISBN 978-0-12-031146-0.
- ↑ Davidson, Peter J.; Harris, David H.; Lappert, Michael F. (1976). "Subvalent Group 4B metal alkyls and amides. Part I. The synthesis and physical properties of kinetically stable bis[bis(trimethysilyl)methyl]-germanium(II), -tin(II), and -lead(II)". Journal of the Chemical Society, Dalton Transactions (21): 2268. doi:10.1039/DT9760002268.
- ↑ Grützmacher, Hansjörg; Freitag, Stefanie; Herbst-Irmer, Regine; Sheldrick, George S. (April 1992). "Investigations of the Structure and Reactivity of a Stannaketenimine". Angewandte Chemie International Edition in English. 31 (4): 437–438. doi:10.1002/anie.199204371.
- ↑ Chen, Xiaodan; Li, Zhongshu; Frenking, Gernot; Fernández, Israel; Zhao, Lili; Grützmacher, Hansjörg (12 June 2019). "Bent Phosphaallenes With "Hidden" Lone Pairs as Ligands". Chemistry – A European Journal. 25 (33): 7912–7920. doi:10.1002/chem.201900645. PMID 30927503. S2CID 88481667.