| ADSL | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | ADSL, AMPS, ASASE, ASL, adenylosuccinate lyase, Adenylosuccinate lyase | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 608222 MGI: 103202 HomoloGene: 12 GeneCards: ADSL | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||



| Adenylosuccinate lyase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
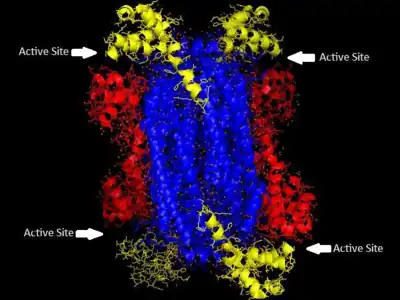 'The homotetrameric structure of ASL in Thermotoga maritima Domain 1 is in red, Domain 2 is in blue, Domain 3 is in yellow. This structure was inspired by a paper by Toth and Yeates[5] | |||||||||
| Identifiers | |||||||||
| EC no. | 4.3.2.2 | ||||||||
| CAS no. | 9027-81-0 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
Adenylosuccinate lyase (or adenylosuccinase) is an enzyme that in humans is encoded by the ADSL gene.[6]
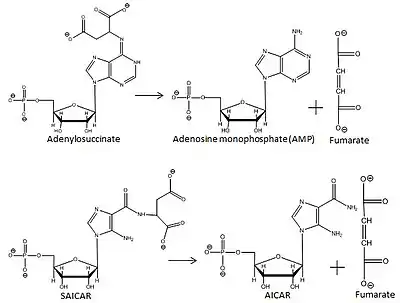
Adenylosuccinate lyase converts adenylosuccinate to AMP and fumarate as part of the purine nucleotide cycle. ASL catalyzes two reactions in the purine biosynthetic pathway that makes AMP; ASL cleaves adenylosuccinate into AMP and fumarate, and cleaves SAICAR into AICAR and fumarate.
Adenylosuccinate lyase is part of the β-elimination superfamily of enzymes and it proceeds through an E1cb reaction mechanism. The enzyme is a homotetramer with three domains in each monomer and four active sites per homotetramer.
Point mutations in adenylosuccinate that cause lowered enzymatic activity cause clinical symptoms that mark the condition adenylosuccinate lyase deficiency.
This protein may use the morpheein model of allosteric regulation.[7]
Function

Adenylosuccinate lyase (ASL) is an enzyme that catalyzes two reactions in the de novo purine biosynthetic pathway. In both reactions it uses an E1cb elimination reaction mechanism to cleave fumarate off of the substrate. In the first reaction, ASL converts 5-aminoimidazole- (N-succinylocarboxamide) ribotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribotide (AICAR) and fumarate. AICAR proceeds through three more reactions before it becomes adenylosuccinate (also called succinyladenosine monophosphate or SAMP), which ASL then splits into adenosine monophosphate (AMP) and fumarate.[8] ASL is important to cells not only because of its involvement in creating purines needed for cellular replication, but also because it helps regulate metabolic processes by controlling the levels of AMP and fumarate in the cell.[9]
Structure
Subunits
Adenylosuccinate lyase belongs to the β-elimination superfamily, and as such its structure is a homotetramer . The monomer of adenylosuccinate lyase has three domains. In Thermotoga maritima, domain 1 contains 7 α-helices in residues 1-93, including the His68 which is highly conserved and was previously thought to be the catalytic acid in the active site.[5] More recent studies have posited that the His171 in domain 2, previously thought to be a catalytic base, may in fact be acting as the catalytic acid, at least in Escherichia coli.[9] Domain 2 is made up of residues 94-341, and contains 5 α-helices and the monomer's only β-sheet. Domain 3 is made up of 7 α-helices. The core of the tetramer is made up of the four domain 2 copies, and there are two copies each of domains 1 and 3 on each end of the tetramer giving the tetramer D2 dihedral symmetry. The tetramer has four active sites, each where three domains meet.[5]
Adenylosuccinate lyase in humans and Bacillus subtilis can be competitively inhibited by the substrate analog adenosine phosphonobutyric acid 2’(3’), 5’-diphosphate (APBADP). APBADP is a competitive inhibitor for both of the reactions catalyzed by adenylosuccinate lyase, and kinetic studies with APBADP show that the substrates for both reactions use the same active site.[10] In the ASL-catalyzed reaction splitting adenylosuccinate into adenosine monophosphate (AMP) and fumarate, the AMP must rotate slightly after the reaction is complete and before fumarate is released in order for both products to fit in the active site.[11]
Mutations
Adenylosuccinate lyase mutants can have considerably reduced activity whether the mutation is in or away from the active site. Disease-causing ASL mutants R396C and R396H are at the entrance to the active site and have lower Vmax than the wild-type ASL, but the mutants K246E and L311V which are away from the active site also cause decreased Vmax. ASL mutant R194C is away from the active site, and though it maintains a Vmax similar to wild-type ASL, it was shown to be the least conformationally stable of the five mutants in vitro and still causes disease.[12]
Mechanism
It was previously thought that the mechanism of action for adenylosuccinate lyase was a concerted catalysis where the hydrogen on the β-carbon (with respect to the leaving nitrogen) was abstracted by the catalytic base at the same time that the leaving nitrogen was protonated by the catalytic acid for E2 elimination.[5] More recent data conflicts with this idea and has confirmed that the mechanism is not in fact concerted, but that the abstraction occurs first and there is an intermediate carbanion species which is resonance stabilized. For both ASL-catalyzed reactions deprotonation of the carbon β to the leaving nitrogen occurs first, then the formation and resonance stabilization of the carbanion occurs, and lastly the protonation of the leaving nitrogen which causes the C-N bond to break.[9] Experimental confirmation of the deprotonation, carbanion formation, and the rate-limiting step of protonation causing cleavage means this is an E1cb mechanism. The most recent data suggest that the catalytic acid is His171, which was previously thought to be the catalytic base, and that somewhat unusually it is a serine at position 295 acts as the catalytic base. The cleavage of adenylosuccinate to AMP and fumarate is an ordered uni-bi mechanism, which means that after cleavage the fumarate leaves the active site before the AMP does.[13]
Role in disease
Mutated adenylosuccinate lyase (ASL) causes clinical disease in patients that is referred to as adenylosuccinate lyase deficiency. This condition is rare, and it presents with varying degrees of psychomotor retardation, autism, muscle wasting, and epilepsy.[14][15] The exact cause of disease is unknown, but possibilities include not enough purine nucleotide synthesis for cell replication, malfunctioning of the purine nucleotide cycle, and a buildup of substrates to toxic levels. Several disease-linked point mutations have been identified, and those who are heterozygous for a point mutation are healthy, but those who are homozygous develop clinical disease.[16] The number of disease-causing genotypes keeps increasing as more mutations are discovered, and now thirty different point mutations have been identified so far, and one deletion, that cause adenylosuccinate lyase deficiency.[17]
When the substrates of ASL (adenylosuccinate and SAICAR) build up due to enzyme deficiency, they are dephosphorylated and turn into succinyladenosine (S-Ado) and succinylaminoimidazole carboximide riboside (SAICA riboside).[18] Normally these compounds are not present in the cerebrospinal fluid or urine because ASL acts on the majority of the substrate molecules before they can build up and be phosphorylated.[15] In the past there has not been a good test for adenylosuccinate lyase deficiency, making the rare disease difficult to diagnose, but recently a test was developed to detect SAICA and S-Ado in the urine. The test is inexpensive and had no false positives or false negatives in the researchers’ small sample.[19]
It is thought that SAICA riboside may be the more toxic compound as it is found at higher levels in patients with severe clinical symptoms, and some researchers think S-Ado may even be protective. More research needs to be done on what determines disease severity, but the instability of human ASL in the lab setting has been an obstacle to this research.[17]
Therapeutic applications
As resistance to anti-malarials increases, researchers are looking for new strategies to target the Plasmodium parasites which cause malaria, especially the more lethal P. falciparum. Some researchers suggested that ASL be looked into as a potential drug target because though interruption of the de novo purine biosynthesis pathway is toxic to the host, Plasmodium ASL has a low level of sequence homology with human ASL which may make any anti-Plasmodium ASL drugs specific enough not to harm human hosts.[20]
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000239900 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000022407 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- 1 2 3 4 5 Toth EA, Yeates TO (February 2000). "The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway". Structure. 8 (2): 163–74. doi:10.1016/S0969-2126(00)00092-7. PMID 10673438.
- ↑ "Entrez Gene: Adenylosuccinate lyase". Retrieved 2012-03-01.
- ↑ Selwood T, Jaffe EK (Mar 2012). "Dynamic dissociating homo-oligomers and the control of protein function". Archives of Biochemistry and Biophysics. 519 (2): 131–43. doi:10.1016/j.abb.2011.11.020. PMC 3298769. PMID 22182754.
- ↑ Spiegel EK, Colman RF, Patterson D (2006). "Adenylosuccinate lyase deficiency". Molecular Genetics and Metabolism. 89 (1–2): 19–31. doi:10.1016/j.ymgme.2006.04.018. PMID 16839792.
- 1 2 3 4 Tsai M, Koo J, Yip P, Colman RF, Segall ML, Howell PL (Jul 2007). "Substrate and product complexes of Escherichia coli adenylosuccinate lyase provide new insights into the enzymatic mechanism". Journal of Molecular Biology. 370 (3): 541–54. doi:10.1016/j.jmb.2007.04.052. PMC 4113493. PMID 17531264.
- ↑ Sivendran S, Colman RF (Jul 2008). "Effect of a new non-cleavable substrate analog on wild-type and serine mutants in the signature sequence of adenylosuccinate lyase of Bacillus subtilis and Homo sapiens". Protein Science. 17 (7): 1162–74. doi:10.1110/ps.034777.108. PMC 2442012. PMID 18469177.
- ↑ Kozlov G, Nguyen L, Pearsall J, Gehring K (Sep 2009). "The structure of phosphate-bound Escherichia coli adenylosuccinate lyase identifies His171 as a catalytic acid". Acta Crystallographica Section F. 65 (Pt 9): 857–61. doi:10.1107/S1744309109029674. PMC 2795585. PMID 19724117.
- ↑ Ariyananda Lde Z, Lee P, Antonopoulos C, Colman RF (Jun 2009). "Biochemical and biophysical analysis of five disease-associated human adenylosuccinate lyase mutants". Biochemistry. 48 (23): 5291–302. doi:10.1021/bi802321m. PMC 2745324. PMID 19405474.
- ↑ Bulusu V, Srinivasan B, Bopanna MP, Balaram H (Apr 2009). "Elucidation of the substrate specificity, kinetic and catalytic mechanism of adenylosuccinate lyase from Plasmodium falciparum". Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1794 (4): 642–54. doi:10.1016/j.bbapap.2008.11.021. PMID 19111634.
- ↑ Maaswinkel-Mooij PD, Laan LA, Onkenhout W, Brouwer OF, Jaeken J, Poorthuis BJ (Aug 1997). "Adenylosuccinase deficiency presenting with epilepsy in early infancy". Journal of Inherited Metabolic Disease. 20 (4): 606–7. doi:10.1023/A:1005323512982. PMID 9266401. S2CID 52833816.
- 1 2 Lee P, Colman RF (Feb 2007). "Expression, purification, and characterization of stable, recombinant human adenylosuccinate lyase". Protein Expression and Purification. 51 (2): 227–34. doi:10.1016/j.pep.2006.07.023. PMID 16973378.
- ↑ Stone RL, Aimi J, Barshop BA, Jaeken J, Van den Berghe G, Zalkin H, Dixon JE (Apr 1992). "A mutation in adenylosuccinate lyase associated with mental retardation and autistic features". Nature Genetics. 1 (1): 59–63. doi:10.1038/ng0492-59. PMID 1302001. S2CID 21577926.
- 1 2 Palenchar JB, Crocco JM, Colman RF (Aug 2003). "The characterization of mutant Bacillus subtilis adenylosuccinate lyases corresponding to severe human adenylosuccinate lyase deficiencies". Protein Science. 12 (8): 1694–705. doi:10.1110/ps.0303903. PMC 2323956. PMID 12876319.
- ↑ Jaeken J, Van den Berghe G (Nov 1984). "An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids". Lancet. 2 (8411): 1058–61. doi:10.1016/s0140-6736(84)91505-8. PMID 6150139. S2CID 54275991.
- ↑ Maddocks J, Reed T (Jan 1989). "Urine test for adenylosuccinase deficiency in autistic children". Lancet. 1 (8630): 158–9. doi:10.1016/S0140-6736(89)91172-0. PMID 2563072. S2CID 1534130.
- ↑ Marshall VM, Coppel RL (Sep 1997). "Characterisation of the gene encoding adenylosuccinate lyase of Plasmodium falciparum". Molecular and Biochemical Parasitology. 88 (1–2): 237–41. doi:10.1016/S0166-6851(97)00054-6. PMID 9274883.
Further reading
- Marie S, Cuppens H, Heuterspreute M, Jaspers M, Tola EZ, Gu XX, Legius E, Vincent MF, Jaeken J, Cassiman JJ, Van den Berghe G (1999). "Mutation analysis in adenylosuccinate lyase deficiency: eight novel mutations in the re-evaluated full ADSL coding sequence". Human Mutation. 13 (3): 197–202. doi:10.1002/(SICI)1098-1004(1999)13:3<197::AID-HUMU3>3.0.CO;2-D. PMID 10090474. S2CID 37296574.
- Kmoch S, Hartmannová H, Stibůrková B, Krijt J, Zikánová M, Sebesta I (Jun 2000). "Human adenylosuccinate lyase (ADSL), cloning and characterization of full-length cDNA and its isoform, gene structure and molecular basis for ADSL deficiency in six patients". Human Molecular Genetics. 9 (10): 1501–13. doi:10.1093/hmg/9.10.1501. PMID 10888601.
- Race V, Marie S, Vincent MF, Van den Berghe G (Sep 2000). "Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency". Human Molecular Genetics. 9 (14): 2159–65. doi:10.1093/hmg/9.14.2159. PMID 10958654.
- Tabucchi A, Carlucci F, Rosi F, Guerranti R, Marinello E (Jun 2001). "Determination, activity and biological role of adenylosuccinate lyase in blood cells". Biomedicine & Pharmacotherapy. 55 (5): 277–83. doi:10.1016/s0753-3322(01)00061-0. PMID 11428554.
- Marie S, Race V, Nassogne MC, Vincent MF, Van den Berghe G (Jul 2002). "Mutation of a nuclear respiratory factor 2 binding site in the 5' untranslated region of the ADSL gene in three patients with adenylosuccinate lyase deficiency". American Journal of Human Genetics. 71 (1): 14–21. doi:10.1086/341036. PMC 384970. PMID 12016589.
- Castro M, Pérez-Cerdá C, Merinero B, García MJ, Bernar J, Gil Nagel A, Torres J, Bermúdez M, Garavito P, Marie S, Vincent F, Van den Berghe G, Ugarte M (Aug 2002). "Screening for adenylosuccinate lyase deficiency: clinical, biochemical and molecular findings in four patients". Neuropediatrics. 33 (4): 186–9. doi:10.1055/s-2002-34493. PMID 12368987. S2CID 20537145.
- Palenchar JB, Colman RF (Feb 2003). "Characterization of a mutant Bacillus subtilis adenylosuccinate lyase equivalent to a mutant enzyme found in human adenylosuccinate lyase deficiency: asparagine 276 plays an important structural role". Biochemistry. 42 (7): 1831–41. doi:10.1021/bi020640+. PMID 12590570.
- Edery P, Chabrier S, Ceballos-Picot I, Marie S, Vincent MF, Tardieu M (Jul 2003). "Intrafamilial variability in the phenotypic expression of adenylosuccinate lyase deficiency: a report on three patients". American Journal of Medical Genetics Part A. 120A (2): 185–90. doi:10.1002/ajmg.a.20176. PMID 12833398. S2CID 21114377.
- Stone RL, Aimi J, Barshop BA, Jaeken J, Van den Berghe G, Zalkin H, Dixon JE (Apr 1992). "A mutation in adenylosuccinate lyase associated with mental retardation and autistic features". Nature Genetics. 1 (1): 59–63. doi:10.1038/ng0492-59. PMID 1302001. S2CID 21577926.
- Sivendran S, Patterson D, Spiegel E, McGown I, Cowley D, Colman RF (Dec 2004). "Two novel mutant human adenylosuccinate lyases (ASLs) associated with autism and characterization of the equivalent mutant Bacillus subtilis ASL". The Journal of Biological Chemistry. 279 (51): 53789–97. doi:10.1074/jbc.M409974200. PMID 15471876.
External links
- Adenylosuccinate+lyase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- Human ADSL genome location and ADSL gene details page in the UCSC Genome Browser.
- Human ASL genome location and ASL gene details page in the UCSC Genome Browser.