Nucleophilic epoxidation is the formation of epoxides from electron-deficient double bonds through the action of nucleophilic oxidants. Nucleophilic epoxidation methods represent a viable alternative to electrophilic methods, many of which do not epoxidize electron-poor double bonds efficiently.[1]
Although the most commonly used asymmetric epoxidation methods (the Sharpless-Katsuki,[2] and Jacobsen[3] epoxidations) rely on the catalytic reactivity of electrophilic oxidants, nucleophilic oxygen sources substituted with a suitable leaving group can also act as epoxidation reagents. The classic example, the Weitz-Scheffer reaction[4] employs hydrogen peroxide under basic conditions (Z = OH below). Other notable examples have employed hypochlorites (Z = Cl) and chiral peroxides (Z = OR*).
(1)

Asymmetric versions of the above reaction have taken advantage of a number of strategies for achieving asymmetric induction. The highest yielding and most enantioselective methods include:
- Use of stoichiometric chiral oxidant[5]
- Use of stoichiometric metal peroxides substituted with chiral ligands[6]
- Use of stoichiometric chiral base[7]
- Use of polypeptides[8]
Although the mechanisms of each of these reactions differ somewhat, in each case the chiral catalyst or reagent must be involved in the enantio determining conjugate addition step. Cis-epoxides are difficult to access using nucleophilic epoxidation methods. Nearly all nucleophilic epoxidations of cis olefins afford trans epoxides.
Mechanism and Stereochemistry
Prevailing Mechanism
The mechanism of nucleophilic epoxidation begins with conjugate addition of the peroxide (or other O-nucleophilic species) to the enone. Metal ions or conjugate acids present in solution coordinate both the peroxide oxygen and the enolate oxygen. Attack of the enolate on the peroxide oxygen generates the epoxide product and releases a leaving group.
(2)
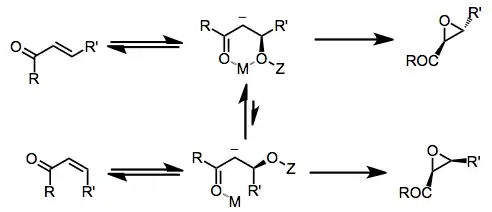
Because the process is stepwise, the configuration of the carbon-carbon double bond is not necessarily preserved. Both cis and trans enones form trans epoxides under nearly all nucleophilic epoxidation conditions (methods employing lanthanide-BINOL systems are the exception).
Stereoselective Variants
How stereoselectivity is achieved in asymmetric nucleophilic epoxidations depends on the method employed. Covered here are various methods for the asymmetric nucleophilic epoxidation of electron-poor olefins. See below for a survey of the substrate scope of the reaction.
When chiral, non-racemic peroxides are used, the two transition states of epoxidation leading to enantiomeric products are diastereomeric. Steric interactions between the peroxide, enone, and templating cation M+ influence the sense of selectivity observed.[9]
(3)
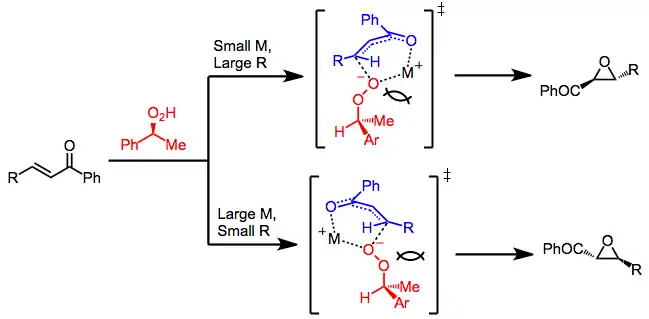
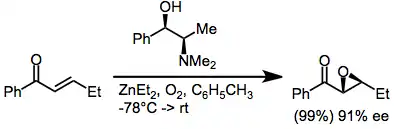
Methods that employ metal peroxides modified by chiral, non-racemic ligands operate by a similar mechanism in which the metal cation plays a templating role. Chiral zinc alkoxides under an oxygen atmosphere have been used to epoxidize some classes of enones (see equation (8) below). The evolution of ethane gas and uptake of oxygen are evidence for ligand exchange followed by oxidation of the intermediate zinc alkoxide species.[10] A catalytic version of this transformation has been achieved using chiral zinc alkylperoxides.[11]
(4)

Lithium, magnesium, and calcium[12] alkylperoxides have also been employed as asymmetric nucleophilic epoxidation reagents. Simple tartrate and pseudoephedrine ligands are effective in combination with these metals; however, little detailed information about the precise mechanisms of these systems is known.
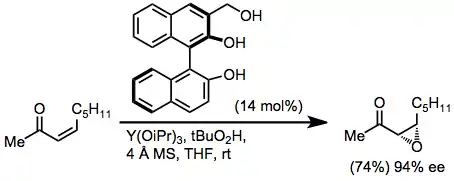
In combination with BINOL ligands and cumene hydroperoxide, lanthanide alkoxides can be used to epoxidize both trans and cis enones with high enantioselectivity. Studies of non-linear effects with these catalyst systems suggest that the active catalyst is oligomeric.[13]
(5)

Homopolymers of amino acids (polypeptides) can also be used to effect enantioselective epoxidations in the presence of an enone and a peroxide. Structure-reactivity relationships have not emerged, but enantioselectivities in these reactions are often high, and polypeptides can often be used when other methods fail.[14]
Phase-transfer catalysis of nucleophilic epoxidation is also possible using cinchona-based alkaloid catalysts. Phase-transfer methods allow some variability in the oxidant used: hydroperoxides, hydrogen peroxide, and hypochlorites have all been used with some success.[15]
Scope and Limitations
Optimal conditions for enantio-selective nucleophilic epoxidation depend on the substrate employed. Although a variety of substrates may be epoxidized using nucleophilic methods, each particular method tends to have limited substrate scope. This section describes asymmetric nucleophilic epoxidation methods, organizing them according to the constitution and configuration of the unsaturated substrate.
Enones
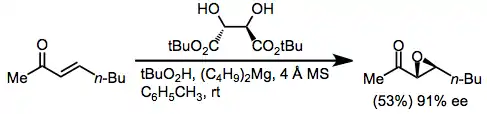
Dialkyl (E)-enones have been most commonly epoxidized using either lanthanide/BINOL systems[16] or a magnesium tartrate catalyst.[17]
(6)
For alkyl aryl (E)-enones, both polypeptides[18] and lanthanide/BINOL catalysts[19] give good yields and enantioselectivities. The most common polypeptide employed is poly-L-leucine.
(7)
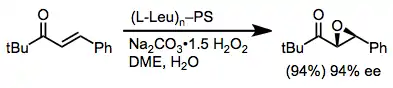
Aryl alkyl (E)-enones have been epoxidized with high enantioselectivity using stoichiometric zinc peroxide systems.[6] Polyleucine may be used with these substrates as well;[18] when an existing stereocenter in the substrate biases the sense of selectivity of the epoxidation, polyleucine is able to overcome this bias.
(8)
Phase-transfer catalysis has been applied successfully to epoxidations of diaryl (E)-enones (chalcones).[15] Lanthanide/BINOL is effective for this class of substrates as well.[20]
(9)

(Z)-Enones are difficult to epoxidize without intermediate bond rotation to afford trans-epoxides. Lanthanide catalysts do effectively prevent bond rotation, however,[19] and provide access to cis epoxide products.
(10)
With the lone exception of methylidene tetralone substrates,[10] no general methods are available for the asymmetric nucleophilic epoxidation of trisubstituted double bonds.
Other Electron-poor Alkenes
Unsaturated esters may be epoxidized using either electrophilic or nucleophilic methods. Lanthanide-mediated epoxidation has been successfully applied to cinnamates and β-heteroaryl unsaturated esters.[21] Amides are also epoxidized under lanthanide-mediated conditions.[22]
(11)

Epoxidations of other electron-deficient double bonds (substituted by electron-withdrawing groups other than carbonyls) are limited in scope, although a few examples have been reported.[23][24] The ability of the carbonyl group to coordinate Lewis acidic functionality is critical for most existing methods.
Comparison with Other Methods
The asymmetric Darzens reaction between aldehydes and (alpha)-haloesters is an effective method for the synthesis of glycidic esters.[25] Chiral auxiliaries,[26] chiral boron enolates,[27] and asymmetric phase transfer catalysis[28] have been used successfully to effect asymmetric induction in the Darzens reaction.
(12)

Diastereoselective epoxidations of chiral, non-racemic alkenes suffer from the limitation that removal of the auxiliary without disturbing the epoxide is often difficult. Nonetheless, diastereoselectivity is high in some cases.[29]
(13)

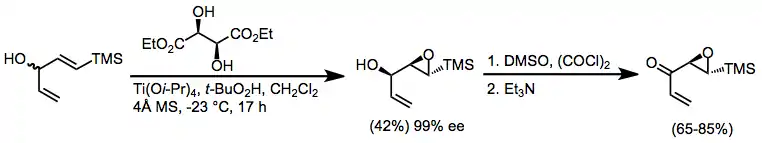
Oxidation of epoxy alcohols generated via Sharpless epoxidation is a third method for the enantioselective synthesis of chiral α,β-epoxy carbonyl compounds.[30] Swern and Parikh-Doering conditions are most commonly applied to accomplish these oxidations.
(14)
Typical experimental conditions
Generally, nucleophilic epoxidations are carried out under inert atmosphere in anhydrous conditions. For zinc-mediated epoxidations, diethylzinc and ligand are first mixed and oxidized, then the enone is introduced. Lanthanide-mediated epoxidations typically require an additive to stabilize the catalyst; this is most commonly triphenylphosphine oxide or triphenylarsine oxide.
Phase-transfer catalyzed epoxidations may be carried out using one of three possible sets of reaction conditions: (1) sodium hypochlorite at room temperature, (2) freshly prepared 8 M potassium hypochlorite, or (3) trichloroisocyanuric acid in aqueous or non-aqueous conditions.
Among polypeptide-based methods, employing a phase transfer catalyst and triphasic media permits lower catalyst loadings. Biphasic conditions using an organic base in conjunction with urea/H2O2 may also be used.
References
- ↑ Porter, M.; Skidmore, J. Org. React. 2009, 74, 425. doi:10.1002/0471264180.or074.03
- ↑ Katsuki, T.; Martin, V. S. Org. React. 1996, 48, 1.
- ↑ Jacobsen, E. N.; Wu, M. H. In Comprehensive Asymmetric Catalysis; Jacobsen, E. N., Pfaltz, A., Yamamoto, H., Eds.; Springer-Verlag: Berlin and Heidelberg, 1999; Vol. II, pp 649–678.
- ↑ Weitz, E.; Scheffer, A. Chem. Ber. 1921, 54, 2327.
- ↑ Adam, W.; Rao, P. B.; Degen, H.-G.; Saha-Möller, C. R. J. Am. Chem. Soc. 2000, 122, 5654.
- 1 2 Enders, D.; Zhu, J. Q.; Raabe, G. Angew. Chem. Int. Ed. Engl. 1996, 35, 1725.
- ↑ Genski, T.; Macdonald, G.; Wei, X.; Lewis, N.; Taylor, R. J. K. Synlett 1999, 795.
- ↑ Da, C. S.; Wei, J.; Dong, S. L.; Xin, Z. Q.; Liu, D. X.; Xu, Z. Q.; Wang, R. Synth. Commun. 2003, 33, 2787.
- ↑ Adam, W.; Rao, P. B.; Degen, H.-G.; Saha-Möller, C. R. Eur. J. Org. Chem. 2002, 630.
- 1 2 Enders, D.; Zhu, J. Q.; Kramps, L. Liebigs Ann./Recl. 1997, 1101.
- ↑ Raheem Keeri, Abdul; Justyniak, Iwona; Jurczak, Janusz; Lewiński, Janusz (2016-03-17). "Quest for Efficient Catalysts based on Zinc tert-Butyl Peroxides for Asymmetric Epoxidation of Enones: C2- vs C1-Symmetric Auxiliaries". Advanced Synthesis & Catalysis. 358 (6): 864–868. doi:10.1002/adsc.201500764. ISSN 1615-4169.
- ↑ Kumaraswamy, G.; Jena, N.; Sastry, M. N. V.; Rao, G. V.; Ankamma, K. J. Mol. Catal. A: Chem. 2005, 230, 59.
- ↑ Daikai, K.; Hayano, T.; Kino, R.; Furuno, H.; Kagawa, T.; Inanaga, J. Chirality 2003, 15, 83.
- ↑ Bickley, J. F.; Hauer, B.; Pena, P. C. A.; Roberts, S. M.; Skidmore, J. J. Chem. Soc., Perkin Trans. 1 2001, 1253.
- 1 2 Corey, E. J.; Zhang, F. Y. Org. Lett. 1999, 1, 1287.
- ↑ Nemoto, T.; Ohshima, T.; Yamaguchi, K.; Shibasaki, M. J. Am. Chem. Soc. 2001, 123, 2725.
- ↑ Jacques, O.; Richards, S. J.; Jackson, R. F. W. Chem. Commun. 2001, 2712.
- 1 2 Allen, J. V.; Drauz, K. H.; Flood, R. W.; Roberts, S. M.; Skidmore, J. Tetrahedron Lett. 1999, 40, 5417.
- 1 2 Watanabe, S.; Kobayashi, Y.; Arai, T.; Sasai, H.; Bougauchi, M.; Shibasaki, M. Tetrahedron Lett. 1998, 39, 7353.
- ↑ Inanaga, J.; Kagawa, T. European Patent 1127616 (2001); Chem. Abstr. 2001, 135, 197209.
- ↑ Kakei, H.; Tsuji, R.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 8962.
- ↑ Nemoto, T.; Kakei, H.; Gnanadesikan, V.; Tosaki, S.-Y.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2002, 124, 14544.
- ↑ Enders, D.; Kramps, L.; Zhu, J. Q. Tetrahedron: Asymmetry 1998, 9, 3959.
- ↑ Geller, T.; Krüger, C. M.; Militzer, H. C. Tetrahedron Lett. 2004, 45, 5069.
- ↑ Newman, M. S.; Magerlein, B. J. Org. React. 1949, 5, 413.
- ↑ Abdel-Magid, A.; Lantos, I.; Pridgen, L. N. Tetrahedron Lett. 1984, 25, 3273.
- ↑ Corey, E. J.; Choi, S. Tetrahedron Lett. 1991, 32, 2857.
- ↑ Arai, S.; Shioiri, T. Tetrahedron Lett. 1998, 39, 2145.
- ↑ Meth-Cohn, W.; Williams, D. J.; Chen, Y. Chem. Commun. 2000, 495.
- ↑ Baldwin, J. E.; Adlington, R. M.; Godfrey, C. R. A.; Patel, V. K. Tetrahedron 1993, 49, 7837.