In bioinformatics, the BLOSUM (BLOcks SUbstitution Matrix) matrix is a substitution matrix used for sequence alignment of proteins. BLOSUM matrices are used to score alignments between evolutionarily divergent protein sequences. They are based on local alignments. BLOSUM matrices were first introduced in a paper by Steven Henikoff and Jorja Henikoff.[1] They scanned the BLOCKS database for very conserved regions of protein families (that do not have gaps in the sequence alignment) and then counted the relative frequencies of amino acids and their substitution probabilities. Then, they calculated a log-odds score for each of the 210 possible substitution pairs of the 20 standard amino acids. All BLOSUM matrices are based on observed alignments; they are not extrapolated from comparisons of closely related proteins like the PAM Matrices.
Biological background
The genetic instructions of every replicating cell in a living organism are contained within its DNA.[2] Throughout the cell's lifetime, this information is transcribed and replicated by cellular mechanisms to produce proteins or to provide instructions for daughter cells during cell division, and the possibility exists that the DNA may be altered during these processes.[2][3] This is known as a mutation. At the molecular level, there are regulatory systems that correct most — but not all — of these changes to the DNA before it is replicated.[3][4]
The functionality of a protein is highly dependent on its structure.[5] Changing a single amino acid in a protein may reduce its ability to carry out this function, or the mutation may even change the function that the protein carries out.[3] Changes like these may severely impact a crucial function in a cell, potentially causing the cell — and in extreme cases, the organism — to die.[6] Conversely, the change may allow the cell to continue functioning albeit differently, and the mutation can be passed on to the organism's offspring. If this change does not result in any significant physical disadvantage to the offspring, the possibility exists that this mutation will persist within the population. The possibility also exists that the change in function becomes advantageous.
The 20 amino acids translated by the genetic code vary greatly by the physical and chemical properties of their side chains.[5] However, these amino acids can be categorised into groups with similar physicochemical properties.[5] Substituting an amino acid with another from the same category is more likely to have a smaller impact on the structure and function of a protein than replacement with an amino acid from a different category.
Sequence alignment is a fundamental research method for modern biology. The most common sequence alignment for protein is to look for similarity between different sequences in order to infer function or establish evolutionary relationships. This helps researchers better understand the origin and function of genes through the nature of homology and conservation. Substitution matrices are utilized in algorithms to calculate the similarity of different sequences of proteins; however, the utility of Dayhoff PAM Matrix has decreased over time due to the requirement of sequences with a similarity more than 85%. In order to fill in this gap, Henikoff and Henikoff introduced BLOSUM (BLOcks SUbstitution Matrix) matrix which led to marked improvements in alignments and in searches using queries from each of the groups of related proteins.[1]
Terminology
- BLOSUM
- Blocks Substitution Matrix, a substitution matrix used for sequence alignment of proteins.
- Scoring metrics (statistical versus biological)
- When evaluating a sequence alignment, one would like to know how meaningful it is. This requires a scoring matrix, or a table of values that describes the probability of a biologically meaningful amino-acid or nucleotide residue-pair occurring in an alignment. Scores for each position are obtained frequencies of substitutions in blocks of local alignments of protein sequences.[7]
- BLOSUM r
- The matrix built from blocks with less than r% of similarity
- E.g., BLOSUM62 is the matrix built using sequences with less than 62% similarity (sequences with ≥ 62% identity were clustered)
- Note: BLOSUM 62 is the default matrix for protein BLAST. Experimentation has shown that the BLOSUM-62 matrix is among the best for detecting most weak protein similarities.[1]
Several sets of BLOSUM matrices exist using different alignment databases, named with numbers. BLOSUM matrices with high numbers are designed for comparing closely related sequences, while those with low numbers are designed for comparing distant related sequences. For example, BLOSUM80 is used for closely related alignments, and BLOSUM45 is used for more distantly related alignments. The matrices were created by merging (clustering) all sequences that were more similar than a given percentage into one single sequence and then comparing those sequences (that were all more divergent than the given percentage value) only; thus reducing the contribution of closely related sequences. The percentage used was appended to the name, giving BLOSUM80 for example where sequences that were more than 80% identical were clustered.
Construction of BLOSUM matrices
BLOSUM matrices are obtained by using blocks of similar amino acid sequences as data, then applying statistical methods to the data to obtain the similarity scores. Statistical Methods Steps : [8]
Eliminating Sequences
Eliminate the sequences that are more than r% identical. There are two ways to eliminate the sequences. It can be done either by removing sequences from the block or just by finding similar sequences and replace them by new sequences which could represent the cluster. Elimination is done to remove protein sequences that are more similar than the specified threshold.
Calculating Frequency & Probability
A database storing the sequence alignments of the most conserved regions of protein families. These alignments are used to derive the BLOSUM matrices. Only the sequences with a percentage of identity lower than the threshold are used. By using the block, counting the pairs of amino acids in each column of the multiple alignment.
Log odds ratio
It gives the ratio of the occurrence each amino acid combination in the observed data to the expected value of occurrence of the pair. It is rounded off and used in the substitution matrix.

where is the probability of observing the pair and is the expected probability of such a pair occurring, given the background probabilities of each amino acid.


BLOSUM Matrices
The odds for relatedness are calculated from log odd ratio, which are then rounded off to get the substitution matrices BLOSUM matrices.
Score of the BLOSUM matrices
A scoring matrix or a table of values is required for evaluating the significance of a sequence alignment, such as describing the probability of a biologically meaningful amino-acid or nucleotide residue-pair occurring in an alignment. Typically, when two nucleotide sequences are being compared, all that is being scored is whether or not two bases are the same at one position. All matches and mismatches are respectively given the same score (typically +1 or +5 for matches, and -1 or -4 for mismatches).[9] But it is different for proteins. Substitution matrices for amino acids are more complicated and implicitly take into account everything that might affect the frequency with which any amino acid is substituted for another. The objective is to provide a relatively heavy penalty for aligning two residues together if they have a low probability of being homologous (correctly aligned by evolutionary descent). Two major forces drive the amino-acid substitution rates away from uniformity: substitutions occur with the different frequencies, and lessen functionally tolerated than others. Thus, substitutions are selected against.[7]
Commonly used substitution matrices include the blocks substitution (BLOSUM) [1] and point accepted mutation (PAM) [10][11] matrices. Both are based on taking sets of high-confidence alignments of many homologous proteins and assessing the frequencies of all substitutions, but they are computed using different methods.[7]
Scores within a BLOSUM are log-odds scores that measure, in an alignment, the logarithm for the ratio of the likelihood of two amino acids appearing with a biological sense and the likelihood of the same amino acids appearing by chance. The matrices are based on the minimum percentage identity of the aligned protein sequence used in calculating them.[12] Every possible identity or substitution is assigned a score based on its observed frequencies in the alignment of related proteins.[13] A positive score is given to the more likely substitutions while a negative score is given to the less likely substitutions.
To calculate a BLOSUM matrix, the following equation is used:

Here, is the probability of two amino acids and replacing each other in a homologous sequence, and and are the background probabilities of finding the amino acids and in any protein sequence. The factor is a scaling factor, set such that the matrix contains easily computable integer values.






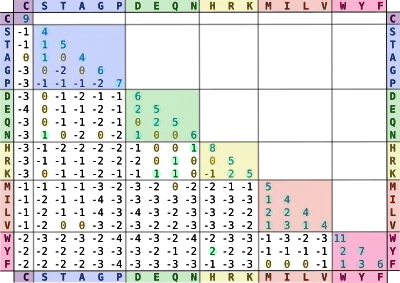
An example - BLOSUM62
BLOSUM80: more related proteins
BLOSUM62: midrange
BLOSUM45: distantly related proteins
An article in Nature Biotechnology[14] revealed that the BLOSUM62 used for so many years as a standard is not exactly accurate according to the algorithm described by Henikoff and Henikoff.[1] Surprisingly, the miscalculated BLOSUM62 improves search performance.[14]
The BLOSUM62 matrix with the amino acids in the table grouped according to the chemistry of the side chain, as in (a). Each value in the matrix is calculated by dividing the frequency of occurrence of the amino acid pair in the BLOCKS database, clustered at the 62% level, divided by the probability that the same two amino acids might align by chance. The ratio is then converted to a logarithm and expressed as a log odds score, as for PAM. BLOSUM matrices are usually scaled in half-bit units. A score of zero indicates that the frequency with which a given two amino acids were found aligned in the database was as expected by chance, while a positive score indicates that the alignment was found more often than by chance, and negative score indicates that the alignment was found less often than by chance.
Some uses in bioinformatics
Research applications
BLOSUM scores was used to predict and understand the surface gene variants among hepatitis B virus carriers[15] and T-cell epitopes.[16]
Surface gene variants among hepatitis B virus carriers
DNA sequences of HBsAg were obtained from 180 patients, in which 51 were chronic HBV carrier and 129 newly diagnosed patients, and compared with consensus sequences built with 168 HBV sequences imported from GenBank. Literature review and BLOSUM scores were used to define potentially altered antigenicity.[15]
Reliable prediction of T-cell epitopes
A novel input representation has been developed consisting of a combination of sparse encoding, Blosum encoding, and input derived from hidden Markov models. this method predicts T-cell epitopes for the genome of hepatitis C virus and discuss possible applications of the prediction method to guide the process of rational vaccine design.[16]
Use in BLAST
BLOSUM matrices are also used as a scoring matrix when comparing DNA sequences or protein sequences to judge the quality of the alignment. This form of scoring system is utilized by a wide range of alignment software including BLAST.[17]
Comparing PAM and BLOSUM
In addition to BLOSUM matrices, a previously developed scoring matrix can be used. This is known as a PAM. The two result in the same scoring outcome, but use differing methodologies. BLOSUM looks directly at mutations in motifs of related sequences while PAM's extrapolate evolutionary information based on closely related sequences.[1]
Since both PAM and BLOSUM are different methods for showing the same scoring information, the two can be compared but due to the very different method of obtaining this score, a PAM100 does not equal a BLOSUM100.[18]
| PAM | BLOSUM |
|---|---|
| PAM100 | BLOSUM90 |
| PAM120 | BLOSUM80 |
| PAM160 | BLOSUM62 |
| PAM200 | BLOSUM50 |
| PAM250 | BLOSUM45 |
The relationship between PAM and BLOSUM
| PAM | BLOSUM |
|---|---|
| To compare closely related sequences, PAM matrices with lower numbers are created. | To compare closely related sequences, BLOSUM matrices with higher numbers are created. |
| To compare distantly related proteins, PAM matrices with high numbers are created. | To compare distantly related proteins, BLOSUM matrices with low numbers are created. |
The differences between PAM and BLOSUM
| PAM | BLOSUM |
|---|---|
| Based on global alignments of closely related proteins. | Based on local alignments. |
| PAM1 is the matrix calculated from comparisons of sequences with no more than 1% divergence but corresponds to 99% sequence identity. | BLOSUM 62 is a matrix calculated from comparisons of sequences with a pairwise identity of no more than 62%. |
| Other PAM matrices are extrapolated from PAM1. | Based on observed alignments; they are not extrapolated from comparisons of closely related proteins. |
| Higher numbers in matrices naming scheme denote larger evolutionary distance. | Larger numbers in matrices naming scheme denote higher sequence similarity and therefore smaller evolutionary distance.[19] |
Software Packages
There are several software packages in different programming languages that allow easy use of Blosum matrices.
Examples are the blosum module for Python, or the BioJava library for Java.
See also
References
- 1 2 3 4 5 6 Henikoff, S.; Henikoff, J.G. (1992). "Amino Acid Substitution Matrices from Protein Blocks". PNAS. 89 (22): 10915–10919. Bibcode:1992PNAS...8910915H. doi:10.1073/pnas.89.22.10915. PMC 50453. PMID 1438297.
- 1 2 Campbell NA; Reece JB; Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). "The Molecular Basis of Inheritance". Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 307–325. ISBN 9781442502215.
- 1 2 3 Campbell NA; Reece JB; Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). "From Gene to Protein". Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 327–350. ISBN 9781442502215.
- ↑ Pal JK, Ghaskadbi SS (2009). "DNA Damage, Repair and Recombination". Fundamentals of Molecular Biology (1st ed.). Oxford University Press. pp. 187–203. ISBN 9780195697810.
- 1 2 3 Campbell NA; Reece JB; Meyers N; Urry LA; Cain ML; Wasserman SA; Minorsky PV; Jackson RB (2009). "The Structure and Function of Large Biological Molecules". Biology: Australian Version (8th ed.). Pearson Education Australia. pp. 68–89. ISBN 9781442502215.
- ↑ Lobo, Ingrid (2008). "Mendelian Ratios and Lethal Genes". Nature. Retrieved 19 October 2013.
- 1 2 3 pertsemlidis A.; Fondon JW.3rd (September 2001). "Having a BLAST with bioinformatics (and avoiding BLASTphemy)". Genome Biology. 2 (10): reviews2002.1–2002.10. doi:10.1186/gb-2001-2-10-reviews2002. PMC 138974. PMID 11597340.
{{cite journal}}: CS1 maint: numeric names: authors list (link) - ↑ "BLOSSUM MATRICES: Introduction to BIOINFORMATICS" (PDF). UNIVERSITI TEKNOLOGI MALAYSIA. 2009. Retrieved 9 September 2014.
- ↑ Murali Sivaramakrishnan; Ognjen Perisic; Shashi Ranjan. "CS#594 - Group 13 (Tools and softwares)" (PDF). University of Illinois at Chicago - UIC. Retrieved 9 September 2014.
- ↑ Margaret O., Dayhoff (1978). "22". Atlas of Protein Sequence and Structure. Vol. 5. Washington DC: National Biomedical Research Foundation. pp. 345–352.
- ↑ States DJ.; Gish W.; Altschul SF. (1991). "Improved sensitivity of nucleic acid database searches using application-specific scoring matrices". Methods: A Companion to Methods in Enzymology. 3: 66–70. CiteSeerX 10.1.1.114.8183. doi:10.1016/s1046-2023(05)80165-3. ISSN 1046-2023.
- ↑ Albert Y. Zomaya (2006). Handbook of Nature-Inspired And Innovative Computing. New York, NY: Springer. ISBN 978-0-387-40532-2.page 673
- ↑ NIH "Scoring Systems"
- 1 2 Mark P Styczynski; Kyle L Jensen; Isidore Rigoutsos; Gregory Stephanopoulos (2008). "BLOSUM62 miscalculations improve search performance". Nat. Biotechnol. 26 (3): 274–275. doi:10.1038/nbt0308-274. PMID 18327232. S2CID 205266180.
- 1 2 Roque-Afonso AM, Ferey MP, Ly TD (2007). "Viral and clinical factors associated with surface gene variants among hepatitis B virus carriers". Antivir Ther. 12 (8): 1255–1263. doi:10.1177/135965350701200801. PMID 18240865. S2CID 9822759.
- 1 2 Nielsen M, Lundegaard C, Worning P, et al. (2003). "Reliable prediction of T‐cell epitopes using neural networks with novel sequence representations" (PDF). Protein Science. 12 (5): 1007–1017. doi:10.1110/ps.0239403. PMC 2323871. PMID 12717023.
- ↑ "The Statistics of Sequence Similarity Scores". National Centre for Biotechnology Information. Retrieved 20 October 2013.
- ↑ Saud, Omama (2009). "PAM and BLOSUM Substitution Matrices". Birec. Archived from the original on 9 March 2013. Retrieved 20 October 2013.
- ↑ "The art of aligning protein sequences Part 1 Matrices". Dai hoc Can Tho - Can Tho University. Archived from the original on 11 September 2014. Retrieved 7 September 2014.
External links
- Sean R. Eddy (2004). "Where did the BLOSUM62 alignment score matrix come from?". Nature Biotechnology. 22 (8): 1035–6. doi:10.1038/nbt0804-1035. PMID 15286655. S2CID 205269887.
- BLOCKS WWW server
- Scoring systems for BLAST at NCBI
- Data files of BLOSUM on the NCBI FTP server.
- Interactive BLOSUM Network Visualization Archived 30 January 2017 at the Wayback Machine