Base-promoted epoxide isomerization is the conversion of alkyl epoxides to ring-opened products through the action of strong base. Isomerizations of this type are most often used to synthesize allylic alcohols, although other products are possible.[1]
Introduction
In the presence of lithium or aluminum amide bases, epoxides may open to give the corresponding allylic alcohols. Removal of a proton adjacent to the epoxide, elimination, and neutralization of the resulting alkoxide lead to synthetically useful allylic alcohol products. In reactions of chiral, non-racemic epoxides, the configuration of the allylic alcohol product matches that of the epoxide substrate at the carbon whose C–O bond does not break (the starred carbon below). Besides β-elimination some other reactions[2][3] are possible, as metalation of the epoxide ring can take place competitively. Vinylogous eliminations are possible when the epoxide substrate is substituted with vinyl or dienyl groups.[4] Unconstrained systems tend to form trans double bonds, as significant non-bonding interactions are avoided in the transition state for the formation of trans products (see equation (2) below). The strongly basic conditions required for most isomerizations of this type represent the reaction's primary disadvantage.
(1)

Mechanism and stereochemistry
Prevailing mechanism
Isomerization of epoxides to allylic alcohols under strongly basic conditions proceeds by a β-elimination process. A model has been advanced that invokes an initial complex between the lithium amide base and epoxide.[5] Concerted C–O bond cleavage and deprotonation proceeds via a syn transition state to give an allylic alkoxide, which is protonated upon workup. Deprotonation typically occurs at the exist in the transition state for cis double bond formation.
(2)

Other processes may take place competitively under basic conditions, particularly when β-elimination is slow or not possible.[6] These pathways likely begin with lithiation of a carbon in the epoxide ring, followed by α-elimination to afford a carbene intermediate. 1,2-hydrogen migration leads to ketones,[2] while intramolecular C–H insertion affords cyclic alcohols with the formation of a new carbon-carbon bond.[3](3)
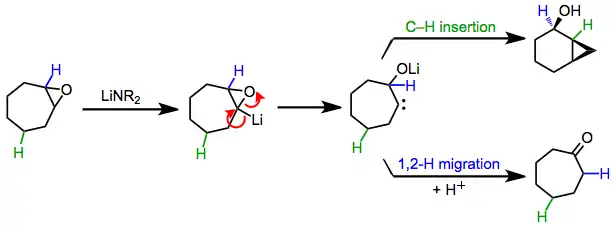
In many cases when hexamethylphosphoramide (HMPA) is used as an additive with lithium amide bases, selectivity for the formation of allylic alcohols increases. These reactions are believed to proceed through E2 elimination.[7]
Stereoselective variants
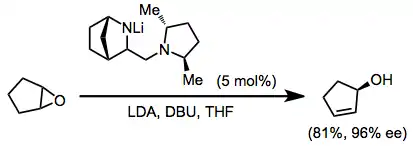
Chiral amide bases may be used in catalytic amounts to isomerize meso epoxides to chiral allylic alcohols with high enantioselectivity.[8]
(4)
Scope and limitations
Terminal epoxides suffer from the limitation that competitive nucleophilic addition of the base to the unsubstituted epoxide carbon may take place. Non-nucleophilic, sterically hindered bases have been used to isomerize terminal epoxides successfully, however.[9](5)

Acyclic disubstituted epoxides undergo deprotonation at the least substituted site (unless a conjugated double bond can be formed; see equation (9) below) with high selectivity for trans double bonds.[10]
(6)

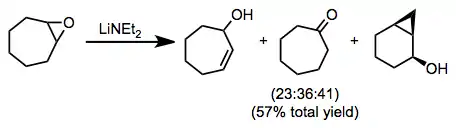
Five- and six-membered rings containing epoxides afford allylic alcohols upon treatment with amide bases; however, reactions of medium-ring epoxides may be complicated by competitive transannular C–H insertion or ketone formation.[11]
(7)
Trisubstituted epoxides do not easily undergo metalation in the ring. As a result, allylic alcohols are formed from these substrates without competitive carbenoid transformations. The use of bulky aluminum amide bases facilitates elimination at the substituent cis to hydrogen, which occurs from the less sterically hindered epoxide-base complex.
(8)

Suitably substituted unsaturated epoxides may undergo vinylogous elimination, which leads to conjugated allylic alcohols. Substituted vinyl epoxides undergo 1,4-elimination in some but not all cases; however, epoxides with β unsaturation eliminate cleanly to give conjugated allylic alcohols.[4]
(9)

Experimental conditions and procedure
Typical conditions
Lithium amides are usually prepared in the laboratory through the addition of a titrated solution of n-butyllithium in hexanes to a solution of the amine in ether. Dry glassware and inert atmosphere are required for these reactions. Alternatively, lithium amides may be prepared by the direct action of lithium on the corresponding amine. Typical temperatures for isomerization reactions employing lithium amides are between 0 °C and reflux (ether/hexane solvent mixtures derived from the synthesis of the lithium amide are usually used directly for isomerization reactions). An excess of the base is employed to account for impurities that consume base and reaction of the base with the ether solvent. Care should be taken when HMPA is added to lithium amide reactions, as it is a known animal carcinogen.
Organolithium reagents may also be used; however, lower temperatures are required to avoid decomposition of the base. These reactions are most often run in hexanes.
Aluminum amides, which are bulkier and sometimes more selective than lithium amides, are prepared from the corresponding lithium amides and diethylaluminum chloride. Reactions are usually carried out at 0 °C in an inert atmosphere, with benzene as the solvent.
Example procedure
(10)

A benzene solution of 1 eq of diethylaluminum chloride was added dropwise at 0° to a solution of 1 eq of lithium 2,2,6,6-tetramethylpiperidide prepared in the usual fashion in benzene. The resulting slurry was stirred for 30 minutes and used immediately. To a stirred mixture of 0.004 mol of diethylaluminum 2,2,6,6-tetramethylpiperidide in 10 mL of benzene at 0 °C was added dropwise over 5 minutes a solution of 0.18 g (0.001 mol) of epoxide in 3 mL of benzene. The mixture was stirred at 0 °C until analysis indicated the absence of starting material. The reaction was quenched by the addition of ice-cold 1 N hydrochloric acid. The organic layer was separated, and the aqueous layer was extracted with ether. The organic layers were combined, washed with brine, dried, and concentrated. The residue was purified by preparative TLC (Rf 0.22 in 1:2 ether–hexane) to give 99% of (E)-2-cyclododecenol: IR (neat) 3330–3370, 1465, 1450, 970 cm−1; NMR (CCl4) δ 3.73–4.20 (1, m), 4.97–5.82 (2, m); mass spectrum (m/z) 182 (16), 164 (13), 139 (32), 125 (46), and 98 (100). [12]
References
- ↑ Crandall, J. K.; Apparu, M. Org. React. 1983, 29, 345. doi:10.1002/0471264180.or029.03
- 1 2 Bond, F. T.; Ho, C. Y. J. Org. Chem. 1976, 41, 1421.
- 1 2 Kirmse, W. Carbene Chemistry, 2nd Ed., Academic Press, New York, 1971, Chapter 7.
- 1 2 Yasuda, A.; Tanaka, S.; Oshima, K.; Yamamoto, H.; Nozaki, H. J. Am. Chem. Soc. 1974, 96, 6513.
- ↑ Sicher, J. Angew. Chem. Int. Ed. Engl. 1972, 11, 200.
- ↑ Cope, C.; Lee, H.; Petree, E. J. Am. Chem. Soc. 1959, 80, 2849.
- ↑ Bartsch, R. A.; Závada, J. Chem. Rev. 1980, 80, 453.
- ↑ Bertillson, S.; Sodergren, M.; Andersson, P. J. Org. Chem. 2002, 67, 1567.
- ↑ Yasuda, A.; Yamamoto, H.; Nozaki, H. Bull. Chem. Soc. Jpn. 1979, 52, 1705.
- ↑ Thummel, P.; Rickborn, B. J. Org. Chem. 1971, 36, 1365.
- ↑ Crandall, J. K.; Chang, H. J. Org. Chem. 1967, 32, 435.
- ↑ Thies, R. W.; Gasic, M.; Whalen, D.; Grutzner, J. D.; Sakai, M.; Johnson, B.; Winstein, S. J. Am. Chem. Soc. 1972, 94, 2262.