CRISPR-Display (CRISP-Disp) is a modification of the CRISPR/Cas9 (Clustered regularly interspaced short palindromic repeats) system for genome editing. The CRISPR/Cas9 system uses a short guide RNA (sgRNA) sequence to direct a Streptococcus pyogenes Cas9 nuclease, acting as a programmable DNA binding protein, to cleave DNA at a site of interest.[1][2]
CRISPR-Display, in contrast, uses a nuclease deficient Cas9 (dCas9) and an engineered sgRNA with aptameric accessory RNA domains, ranging from 100bp to 5kb, outside of the normal complementary targeting sequence.[3] The accessory RNA domains can be functional domains, such as long non-coding RNAs (lncRNAs), protein-binding motifs, or epitope tags for immunochemistry. This allows for investigation of the functionality of certain lncRNAs, and targeting of ribonucleoprotein (RNP) complexes to genomic loci.
CRISPR-Display was first published in Nature Methods in July 2015, and developed by David M. Shechner, Ezgi Hacisuleyman, Scott T. Younger and John Rinn at Harvard University and Massachusetts Institute of Technology (MIT), USA.
Background
The CRISPR/Cas9 system is based on an adaptive immune system of prokaryotic organisms, and its use for genome editing was first proposed and developed in collaboration between Jennifer Doudna (University of California, Berkeley) and Emmanuelle Charpentier (Max Planck Institute for Infection Biology, Germany).[1] The method, and its application in editing human cells, was published in Science on August 17, 2012. In January 2013, the Feng Zhang lab at the Broad Institute at MIT published another method in Science, having further optimized the sgRNA structure and expression for use in mammalian cells.[2] By the beginning of 2014, almost 2500 studies mentioning CRISPR in their title has been published.
Non-coding RNAs (ncRNAs) are RNA transcripts that are not translated into a protein product, but instead exert their function as RNA molecules. They are involved in a range of processes, like post-transcriptional regulation of gene expression, genomic imprinting, and regulating the chromatin state, and thereby the expression, of a given locus. Many ncRNAs have been discovered, but in many cases, their function has yet to be accurately dissected due to technical challenges. ncRNA function is often not affected by introducing point mutations and premature stop codons. ncRNAs are also thought to regulate gene expression, so deletion studies have a hard time distinguishing effects of ncRNA loss from effects of gene misregulation due to the deletion. Studies of ncRNAs have also lacked the throughput necessary for discerning the RNA based functionality. To meet these challenges, the Rinn lab therefore developed a synthetic biology approach, using CRISPR/Cas9 system, with the Cas9 acting as a conduit, to target ncRNA modules to ectopic genomic locations, and investigating the ncRNAs effects on reporter genes and other genomic features at that site.
Method development
CRISPR-Disp modifies the CRISPR/Cas9 technology by using a catalytically inactive, i.e. nuclease deficient, Cas9 mutant (dCas9), and altering the RNA used for targeting Cas9 to a genomic location.
Since sgRNAs are usually expressed by RNA polymerase III, which limits the length of the RNA domain that can be inserted, CRISPR-Display incorporates RNA polymerase II to permit expression of longer transcripts (~80–250 nucleotides) to overcome this limitation. CRISPR-Display can therefore add larger RNA domains, like natural and lncRNA domains, without affecting dCas9 localization.
The sgRNA is engineered with an aptameric accessory RNA domain in the sequence outside of the targeting sequence. In the development of the technique, five model cofactors with different topology constructs were used: TOP1-4 and INT with an accessory domain (P4-P6 domain) at different positions, including the 5’ and 3’ end and internally within the sgRNA. Each domain contained a stem-loop that can be recognized by a PP7 bacteriophage coat protein. The complex was delivered into mammalian cells (HEK293FT cells) by a lentiviral vector.
To ensure that the attached RNA module both retains targeting functionality as well as the resulting complex drive transcriptional activation at a specific site of interest, transient reporter gene expression of luciferase and fluorescent protein was measured. Two variations of such a transcription activator assay was performed; directly with a dCas9 fused to a transcriptional activator/repressor (VP64, a factor known to enhance gene expression) (Direct activation) or indirectly where the transcriptional activator is fused to an RNA binding protein module on the sgRNA (Bridged activation). Reporter gene activation through direct activation imply the sgRNA variant binds and targets dCas9 efficiently. All the five topologies showed direct activation except TOP3 and TOP4, which showed reduced activity. Bridged activation indicates that the fused RNA accessory domain is intact in mature dCas9 complexes. Bridged activation was observed with TOP1, TOP3 and INT. The results were recapitulated at endogenous loci by targeting minimal sgRNA and selective expanded topologies (TOP1 and INT) to human ASCL1, IL1RN, NTF3 and TTN promoters. Direct and bridged activation were observed by qRT-PCR for each construct proving that CRISP-Disp allows deployment of large RNA domains to genomic loci.
The effect of internal (stem-loop) insertion size on dCas9 complex was assessed using INT-like constructs with cassettes of PP7 stem loops with a size range from 25 nt to 247 nt. Each construct induced significant activation in the reporter assays signifying that internal insertion size does not influence the dCas9 complex function. Similarly, the effect of internal insert sequence was also determined through a set of unique sgRNA variants displaying cassettes of 25 random nucleotides. Reporter assays and RIP-Seq confirmed that sequence does not govern complex efficacy.
The utility of CRISP-Disp was explored with an array of functional RNA domains such as natural protein binding motifs, artificial aptamers and small molecules with varying size. While all the complexes were functional and viable, and successfully deployed the RNA domains at endogenous loci, the efficacy changed with length and expression levels. This suggests that optimization of structure and sequence might be important required before designing the construct.
To determine if artificial lncRNA scaffolds can be used with CRISPR-Display, dCas9 complexes were assembled with artificial RNA with a size comparable to lncRNAs. The constructs were expanded to ~650nt size with an additional P4-P6 domain with hairpin loops that can be recognized by another phage coat protein, MS2. These topology constructs were called double TOP0-2 with the two domains either together at 5’ or 3’end or separately at each end. Transient reporter assays followed by confirmation with RNA Immunoprecipitation sequencing (RIP-qPCR) showed that all the three constructs retained both the domains in the complex.
This was also tested with natural lncRNA domains by building Pol II-driven TOP1 and INT constructs fused with human lncRNA domains. lncRNAs used had lengths between ~90–4800 nt, and included the NoRC-binding pRNA, three enhancer-transcribed RNAs (eRNAs) FALEC, TRERNA1 and ncRNA-a3), Xist A-repeat (RepA), and the 4,799-nt transcriptional activator HOTTIP. While all the constructs showed significant direct activation, it decreased with increasing lncRNA-sgRNA length. These lncRNA domains could regulate the reporters independent of dCas9 with pRNA and RepA repressing the GLuc reporter expression (repressors) and TRERNA1, ncRNA-a3 and HOTTIP inducing activation (activators), but were properly targeted to an ectopic location of interest by using the CRISP-Disp system.[3]
Thus, CRISP-Disp enables control of gene expression with deployment of both artificial scaffolds as well as natural lncRNA domains.
Applications
CRISPR-Display allows for previously unavailable studies of lncRNA functionality, artificial ncRNA functionalization, recruitment of endogenous and engineered proteins to genomic loci, and locus affinity tagging for cell imaging.
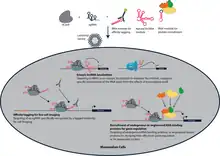
lncRNA domain localization
CRISPR-Display allows targeted localization of natural lncRNAs to ectopic sites for investigation of their function. Exposing various ectopic DNA loci to natural lncRNAs can help show the effects of lncRNAs on gene expression and chromatin state, and help dissect the mechanism of such effects. One of the major outstanding questions in the study of lncRNAs is whether effects on chromatin state or gene expression adjacent to a lncRNA locus is due to functional, sequence-specific mechanisms of the lncRNA itself, or due simply to the act of transcribing the lncRNA.[4][5] Localizing lncRNA to ectopic sites with CRISPR-Display can help separate the function of the RNA itself from the effects of transcribing such RNA species. Before CRISPR-Display, such studies were challenging due to low throughput, and inability to distinguish lncRNA function from other confounding factors like cryptically encoded peptides or functional DNA elements.
Artificial ncRNA functionalization
CRISPR-Display also allows for targeted use of the wide array of artificial RNAs with specific functionality, such as RNAs for recruitment of endogenous RNA-binding proteins, antibody affinity tagging, and recruitment of tagged fusion proteins.
Affinity tagging for live cell imaging
One example of artificial ncRNA functionalization is incorporating RNA domains recognized by specific antibodies to the sgRNA. CRISPR-Display can target the sgRNA with a particular epitope sequence to various loci, and fluorescently tagged antibodies can be used to image the locus, showing its localization in the nucleus, and possible interactions with other tagged proteins or genomic loci.
Recruitment of endogenous or engineered RNA binding proteins for gene regulation
Endogenous proteins known to bind a specific RNA motif can be recruited to ectopic genomic locations by incorporating the RNA motif into the sgRNA. CRISPR-Display can also recruit fusion proteins engineered to bind specific RNA sequences. Recruiting these proteins can allow studies of specific proteins’ and protein complexes’ effects on gene regulation and chromatin states, as well as specific regulation of certain genes for investigation of gene function.
Multiplexed functional studies
Due to the modularity of the sgRNA, several different sgRNAs with distinct functional modules can be expressed in each cell at once. The different RNA modules can then work simultaneously and independently, allowing for, for example, regulation of one genomic location whilst imaging the effects of the regulation at another location.
The possible applications of CRISPR-Display will continue to increase with further development and understanding of ncRNA functionalization. It is not unreasonable to think that CRISPR-Display may one day enable complex synthetic biology systems, with distinct temporal expression of sgRNAs, and networks and circuits of gene regulation by targeting of regulatory proteins.
Advantages
- Can easily accommodate large RNA cargo (up to ~4.8kb, possibly even larger) within the sgRNA core. Therefore, structured RNA domains, natural long lncRNAs, artificial RNA modules and pools of random sequences can be used with dCas9.
- sgRNA-dCas9 complexation is not limited by sequence composition of the RNA cargo, but seems independent of the RNA modules used.
- CRISPR-Display is a modular method, which allows different functions to be simultaneously performed at diverse loci in the same cell. Using a single construct with orthogonal RNA binding proteins where each protein is fused to a unique functional domain and targeted by sgRNA containing its related RNA motif. (multiplexing) (already included in the applications)
- RNA modules can be added at different locations within the sgRNA sequence (internally, or at the 5’ or 3’ ends), so the location of the RNA module can be optimized or best function.
- Allows construction of Cas9 complexes with protein binding cassettes, artificial aptamers, pools of random sequences as well as natural lncRNAs.
Limitations
- CRISPR-Display is currently limited by the number of available functional RNA motifs and RNA binding protein functions. As more such motifs and proteins are discovered and developed, further applications of CRISPR-Display may become possible.
- dCas9 complexation decreases with increasing sgRNA-lncRNA length. The quantitative yields of intact lncRNA domains are, however, recovered relative to the respective sgRNA. Therefore, construct integrity can depend on factors like length and RNA structure.
- Design of a high efficiency CRISPR-Display construct may require some structural or sequence optimization, which can lead to variable construct efficacy.
References
- 1 2 Jinek, Martin; Chylinski, Krzysztof; Fonfara, Ines; Hauer, Michael; Doudna, Jennifer A.; Charpentier, Emmanuelle (2012-08-17). "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity". Science. 337 (6096): 816–821. Bibcode:2012Sci...337..816J. doi:10.1126/science.1225829. ISSN 1095-9203. PMC 6286148. PMID 22745249.
- 1 2 Cong, Le; Ran, F. Ann; Cox, David; Lin, Shuailiang; Barretto, Robert; Habib, Naomi; Hsu, Patrick D.; Wu, Xuebing; Jiang, Wenyan (2013-02-15). "Multiplex genome engineering using CRISPR/Cas systems". Science. 339 (6121): 819–823. Bibcode:2013Sci...339..819C. doi:10.1126/science.1231143. ISSN 1095-9203. PMC 3795411. PMID 23287718.
- 1 2 Shechner, David M.; Hacisuleyman, Ezgi; Younger, Scott T.; Rinn, John L. (2015-07-01). "Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display". Nature Methods. 12 (7): 664–670. doi:10.1038/nmeth.3433. ISSN 1548-7105. PMC 4821475. PMID 26030444.
- ↑ Perez-Pinera, Pablo; Jones, Matthew F.; Lal, Ashish; Lu, Timothy K. (2015-07-16). "Putting Non-coding RNA on Display with CRISPR". Molecular Cell. 59 (2): 146–148. doi:10.1016/j.molcel.2015.07.002. ISSN 1097-4164. PMC 6314022. PMID 26186289.
- ↑ Melé, Marta; Rinn, John L. (2016-06-02). ""Cat's Cradling" the 3D Genome by the Act of LncRNA Transcription". Molecular Cell. 62 (5): 657–664. doi:10.1016/j.molcel.2016.05.011. ISSN 1097-4164. PMID 27259198.