Cardiac excitation-contraction coupling (Cardiac EC coupling) describes the series of events, from the production of an electrical impulse (action potential) to the contraction of muscles in the heart.[1] This process is of vital importance as it allows for the heart to beat in a controlled manner, without the need for conscious input. EC coupling results in the sequential contraction of the heart muscles that allows blood to be pumped, first to the lungs (pulmonary circulation) and then around the rest of the body (systemic circulation) at a rate between 60 and 100 beats every minute, when the body is at rest.[2] This rate can be altered, however, by nerves that work to either increase heart rate (sympathetic nerves) or decrease it (parasympathetic nerves), as the body's oxygen demands change. Ultimately, muscle contraction revolves around a charged atom (ion), calcium (Ca2+),[3] which is responsible for converting the electrical energy of the action potential into mechanical energy (contraction) of the muscle. This is achieved in a region of the muscle cell, called the transverse tubule during a process known as calcium induced calcium release.[4]
Initiation
Located in the wall of the right atrium is a group of specialised cells, called the Sinoatrial node (SAN). These cells, unlike most other cells within the heart, can spontaneously produce action potentials.[5] These action potentials travel along the cell membrane (sarcolemma), as impulses, passing from one cell to the next through channels, in structures known as gap junctions.[6] The speed of conduction of the action potential varies at different parts of the heart (for more information, see electrical conduction system of the heart). This is important as it means that once the atria have contracted, there is a slight delay which enables the ventricles to fill with blood before they contract.
Calcium-induced calcium release
Certain regions of the sarcolemma penetrate deep into the cell. These are known as transverse-tubules (t-tubules); which are also found in skeletal muscle cells and allow for the action potential to travel into the centre of the cell.[7] Special proteins called L-type calcium channels (also known as dihydropyridine receptors (DHPR)) are located on the t-tubule membrane, and are activated by the action potential. Activated DHPRs open, forming a channel, that allows Ca2+ to pass into the cell. This increase in Ca2+, then binds to and activates another receptor, called a type 2 ryanodine receptor (RyR2), located on the membrane of a structure known as the sarcoplasmic reticulum (SR). The SR is a Ca2+ store within the cell and is located very close to the T-tubule. Activation of RyR2 causes it to open, releasing even more Ca2+ into the cell, this release of calcium is called a calcium spark. This means that the initial flow of Ca2+ into the cell, caused a larger release of Ca2+ within the cell, therefore the process is called calcium induced calcium release (CICR).[8]
Muscle Contraction
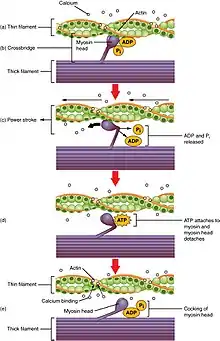
The increase in Ca2+, produced by CICR, now does two things. Firstly, it binds to the intracellular side of the DHPR, signalling the channels to close and preventing further influx of Ca2+ into the cell. Secondly Ca2+ indirectly activates proteins, called myofilaments, resulting in muscle contraction. The two main myofilaments in cardiac (and skeletal) muscle are actin and myosin. Ca2+ binds to a protein called troponin, which is bound to the actin filament. This binding causes a shape change in the troponin which exposes areas on the actin, to which the head of the myosin filament binds. The binding of the myosin head to actin is known as a cross-bridge. A molecule, called adenosine triphosphate (ATP) which is produced by an intracellular structure called a mitochondrion, is then used, as a source of energy, to help move the myosin head, carrying the actin. As a result, the actin slides across the myosin filament shortening the muscle. This is called a power stroke. Myosin then detaches from the actin and resets itself back to its original position, binding to another part of the actin and producing another power stroke, shortening the muscle further. This process continues, with the myosin head moving in a motion similar to that of an oar rowing a boat, until the Ca2+ level within the cell decreases (see figure 1).[9]
Termination of contraction
Contraction ends when the Ca2+ is removed from the cell. When this happens, the troponin changes back to its original shape, blocking the binding sites on actin and preventing the formation of crossbridges. This decrease in Ca2+ within the cell is brought about by a variety of proteins, known collectively as ion transporters. The main pumps involved are: the sarcoplasmic reticulum Ca2+-ATPase, which pumps Ca2+ back into the SR, the Sarcolemmal sodium-calcium exchanger, which pumps one Ca2+ out of the cell, in exchange for 3 sodium ions being pumped into the cell, the Sarcolemmal Ca2+-ATPase, which uses ATP to pump Ca2+ directly out of the cell and the Mitochondrial Ca2+ Uniport system, which pumps Ca2+ into the mitochondria.[10]
Heart rate
Heart rate is affected by nerves. Sympathetic nerves, coming from the spinal cord, increase heart rate, whereas parasympathetic nerves (branching from the vagus nerves) work to decrease it.
Sympathetic nerves work by releasing a protein (neurotransmitter) called noradrenaline which binds to a specific receptor (beta 1 adrenoceptor) located in the sarcolemma and the t-tubule membrane of cardiac cells. This activates a protein, called a G-protein and results in a series of reactions (known as a cyclic AMP pathway) that leads to the production of a molecule called cAMP (cyclic adenosine monophosphate). In the SAN cAMP binds to an ion channel involved in action potential initiation, speeding up the production of the action potential (see sinoatrial node for more detail). cAMP also, activates a protein called protein kinase A (PKA). PKA affects both the L-type calcium channels (also known as dihydropyridine receptors (DHPR)) and RyR, increasing the rise in Ca 2+ within the contractile cells and therefore increasing rate of muscle contraction. PKA also affects the myofilaments as well as a protein called phospholamban (PLB; see sarcoplasmic reticulum for more details), speeding up the rate of Ca2+decline in the cell and so speeding up muscle relaxation.[11]
Parasympathetic nerves work by releasing a neurotransmitter called acetylcholine (ACh) which binds to specific receptor (M2 muscarinic receptor) on the sarcolemma of both SAN cells and ventricular cells. This again activates a G-protein. However this G-protein works by inhibiting, the cAMP pathway, therefore, preventing the sympathetic nervous system from increasing heart rate. As well as this, in the SAN, the G-protein activates specific potassium channel, that opposes action potential initiation (see SAN for more details), thus slowing heart rate.[11]
References
- ↑ Santana, L.F., Cheng, E.P. and Lederer, J.W. (2010) 'How does the shape of the cardiac action potential control calcium signaling and contraction in the heart?', 49(6).
- ↑ Gordan, R., Gwathmey, J.K. and Xie, L.-H. (2015) 'Autonomic and endocrine control of cardiovascular function', 7(4).
- ↑ Marks, A.R. (2003) 'Calcium and the heart: A question of life and death', 111(5).
- ↑ Wong, A.Y.K., Fabiato, A. and Bassingthwaigthe, J.B. (1992) 'MODEL OF CALCIUM-INDUCED CALCIUM RELEASE MECHANISM IN CARDIAC CELLS', 54(1).
- ↑ MONFREDI, O., DOBRZYNSKI, H., MONDAL, T., BOYETT, M.R. and MORRIS, G.M. (2010) 'The anatomy and physiology of the Sinoatrial Node-A contemporary review', Pacing and Clinical Electrophysiology, 33(11), pp. 1392–1406. doi: 10.1111/j.1540-8159.2010.02838.x.
- ↑ Kurtenbach, S. and Zoidl, G. (2014) 'Gap junction modulation and its implications for heart function', 5.
- ↑ Hong, T., Shaw, R.M., Institute, C.-S.H., Center, C.-S.M., Angeles, L., California and Angeles, C.L. (2017) 'Cardiac T-Tubule Microanatomy and function', Reviews, 97(1), pp. 227–252. doi: 10.1152/physrev.00037.2015.
- ↑ Hinch, R., Greenstein, J.L., Tanskanen, A.J., Xu, L. and Winslow, R.L. (2004) 'A simplified local control model of calcium-induced calcium release in cardiac ventricular Myocytes', 87(6).
- ↑ Lodish, H., Berk, A., Zipursky, L.S., Matsudaira, P., Baltimore, D. and Darnell, J. (2000a) Muscle: A specialized Contractile machine. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21670/ (Accessed: 13 February 2017).
- ↑ Balke, C.W., Egan, T.M. and Wier, W.G. (1994) 'Processes that remove calcium from the cytoplasm during excitation-contraction coupling in intact rat heart cells', 474(3).
- 1 2 Gordan, R., Gwathmey, J.K. and Xie, L.-H. (2015b) 'Autonomic and endocrine control of cardi-ovascular function', 7(4).