
The cardiac action potential is a brief change in voltage (membrane potential) across the cell membrane of heart cells.[1] This is caused by the movement of charged atoms (called ions) between the inside and outside of the cell, through proteins called ion channels. The cardiac action potential differs from action potentials found in other types of electrically excitable cells, such as nerves. Action potentials also vary within the heart; this is due to the presence of different ion channels in different cells.
Unlike the action potential in skeletal muscle cells, the cardiac action potential is not initiated by nervous activity. Instead, it arises from a group of specialized cells known as pacemaker cells, that have automatic action potential generation capability. In healthy hearts, these cells form the cardiac pacemaker and are found in the sinoatrial node in the right atrium. They produce roughly 60–100 action potentials every minute. The action potential passes along the cell membrane causing the cell to contract, therefore the activity of the sinoatrial node results in a resting heart rate of roughly 60–100 beats per minute. All cardiac muscle cells are electrically linked to one another, by intercalated discs which allow the action potential to pass from one cell to the next.[2][3] This means that all atrial cells can contract together, and then all ventricular cells.

Rate dependence of the action potential is a fundamental property of cardiac cells and alterations can lead to severe cardiac diseases including cardiac arrhythmia and sometimes sudden death.[4] Action potential activity within the heart can be recorded to produce an electrocardiogram (ECG). This is a series of upward and downward spikes (labelled P, Q, R, S and T) that represent the depolarization (voltage becoming more positive) and repolarization (voltage becoming more negative) of the action potential in the atria and ventricles.[5]
Overview
| Element | Ion | Extracellular | Intracellular | Ratio |
|---|---|---|---|---|
| Sodium | Na+ | 135 - 145 | 10 | 14:1 |
| Potassium | K+ | 3.5 - 5.0 | 155 | 1:30 |
| Chloride | Cl− | 95 - 110 | 10 - 20 | 4:1 |
| Calcium | Ca2+ | 2 | 10−4 | 2 x 104:1 |
| Although intracellular Ca2+ content is about 2 mM, most of this is bound or sequestered in intracellular organelles (mitochondria and sarcoplasmic reticulum).[6] | ||||
Similar to skeletal muscle, the resting membrane potential (voltage when the cell is not electrically excited) of ventricular cells is around −90 millivolts (mV; 1 mV = 0.001 V), i.e. the inside of the membrane is more negative than the outside. The main ions found outside the cell at rest are sodium (Na+), and chloride (Cl−), whereas inside the cell it is mainly potassium (K+).[7]
The action potential begins with the voltage becoming more positive; this is known as depolarization and is mainly due to the opening of sodium channels that allow Na+ to flow into the cell. After a delay (known as the absolute refractory period), the action potential terminates as potassium channels open, allowing K+ to leave the cell and causing the membrane potential to return to negative, this is known as repolarization. Another important ion is calcium (Ca2+), which can be found inside the cell in the sarcoplasmic reticulum (SR) where calcium is stored, and is also found outside of the cell. Release of Ca2+ from the SR, via a process called calcium-induced calcium release, is vital for the plateau phase of the action potential (see phase 2, below) and is a fundamental step in cardiac excitation-contraction coupling.[8]
There are important physiological differences between the pacemaker cells of the sinoatrial node, that spontaneously generate the cardiac action potential and those non-pacemaker cells that simply conduct it, such as ventricular myocytes). The specific differences in the types of ion channels expressed and mechanisms by which they are activated results in differences in the configuration of the action potential waveform, as shown in figure 2.
Cardiac automaticity
Cardiac automaticity also known as autorhythmicity, is the property of the specialized conductive muscle cells of the heart to generate spontaneous cardiac action potentials.[9][10] Automaticity can be normal or abnormal, caused by temporary ion channel characteristic changes such as certain medication usage, or in the case of abnormal automaticity the changes are in electrotonic environment, caused, for example, by myocardial infarction.[11]
Phases

The standard model used to understand the cardiac action potential is that of the ventricular myocyte. Outlined below are the five phases of the ventricular myocyte action potential, with reference also to the SAN action potential.

Phase 4
In the ventricular myocyte, phase 4 occurs when the cell is at rest, in a period known as diastole. In the standard non-pacemaker cell the voltage during this phase is more or less constant, at roughly -90 mV.[12] The resting membrane potential results from the flux of ions having flowed into the cell (e.g. sodium and calcium), the flux of ions having flowed out of the cell (e.g. potassium, chloride and bicarbonate), as well as the flux of ions generated by the different membrane pumps, being perfectly balanced.
The activity of these pumps serve two purposes. The first is to maintain the existence of the resting membrane potential by countering the depolarisation due to the leakage of ions not at the electrochemical equilibrium (e.g. sodium and calcium). These ions not being at the equilibrium is the reason for the existence of an electrical gradient, for they represent a net displacement of charges across the membrane, which are unable to immediately re-enter the cell to restore the electrical equilibrium. Therefore, their slow re-entrance in the cell needs to be counterbalanced or the cell would slowly lose its membrane potential.
The second purpose, intricately linked to the first, is to keep the intracellular concentration more or less constant, and in this case to re-establish the original chemical gradients, that is to force the sodium and calcium which previously flowed into the cell out of it, and the potassium which previously flowed out of the cell back into it (though as the potassium is mostly at the electrochemical equilibrium, its chemical gradient will naturally reequilibrate itself opposite to the electrical gradient, without the need for an active transport mechanism).
For example, the sodium (Na+) and potassium (K+) ions are maintained by the sodium-potassium pump which uses energy (in the form of adenosine triphosphate (ATP)) to move three Na+ out of the cell and two K+ into the cell. Another example is the sodium-calcium exchanger which removes one Ca2+ from the cell for three Na+ into the cell.[13]
During this phase the membrane is most permeable to K+, which can travel into or out of cell through leak channels, including the inwardly rectifying potassium channel.[14] Therefore, the resting membrane potential is mostly equal to K+ equilibrium potential and can be calculated using the Goldman-Hodgkin-Katz voltage equation.
However, pacemaker cells are never at rest. In these cells, phase 4 is also known as the pacemaker potential. During this phase, the membrane potential slowly becomes more positive, until it reaches a set value (around -40 mV; known as the threshold potential) or until it is depolarized by another action potential, coming from a neighboring cell.
The pacemaker potential is thought to be due to a group of channels, referred to as HCN channels (Hyperpolarization-activated cyclic nucleotide-gated). These channels open at very negative voltages (i.e. immediately after phase 3 of the previous action potential; see below) and allow the passage of both K+ and Na+ into the cell. Due to their unusual property of being activated by very negative membrane potentials, the movement of ions through the HCN channels is referred to as the funny current (see below).[15]
Another hypothesis regarding the pacemaker potential is the 'calcium clock'. Calcium is released from the sarcoplasmic reticulum within the cell. This calcium then increases activation of the sodium-calcium exchanger resulting in the increase in membrane potential (as a +3 charge is being brought into the cell (by the 3Na+) but only a +2 charge is leaving the cell (by the Ca2+) therefore there is a net charge of +1 entering the cell). This calcium is then pumped back into the cell and back into the SR via calcium pumps (including the SERCA).[16]
Phase 0
This phase consists of a rapid, positive change in voltage across the cell membrane (depolarization) lasting less than 2 ms in ventricular cells and 10–20 ms in SAN cells.[17] This occurs due to a net flow of positive charge into the cell.
In non-pacemaker cells (i.e. ventricular cells), this is produced predominantly by the activation of Na+ channels, which increases the membrane conductance (flow) of Na+ (gNa). These channels are activated when an action potential arrives from a neighbouring cell, through gap junctions. When this happens, the voltage within the cell increases slightly. If this increased voltage reaches the threshold potential (approximately −70 mV) it causes the Na+ channels to open. This produces a larger influx of sodium into the cell, rapidly increasing the voltage further to around +50 mV,[7] i.e. towards the Na+ equilibrium potential. However, if the initial stimulus is not strong enough, and the threshold potential is not reached, the rapid sodium channels will not be activated and an action potential will not be produced; this is known as the all-or-none law.[18][19] The influx of calcium ions (Ca2+) through L-type calcium channels also constitutes a minor part of the depolarisation effect.[20] The slope of phase 0 on the action potential waveform (see figure 2) represents the maximum rate of voltage change of the cardiac action potential and is known as dV/dtmax.
In pacemaker cells (e.g. sinoatrial node cells), however, the increase in membrane voltage is mainly due to activation of L-type calcium channels. These channels are also activated by an increase in voltage, however this time it is either due to the pacemaker potential (phase 4) or an oncoming action potential. The L-type calcium channels are activated more slowly than the sodium channels, therefore, the depolarization slope in the pacemaker action potential waveform is less steep than that in the non-pacemaker action potential waveform.[12][21]
Phase 1
This phase begins with the rapid inactivation of the Na+ channels by the inner gate (inactivation gate), reducing the movement of sodium into the cell. At the same time potassium channels (called Ito1) open and close rapidly, allowing for a brief flow of potassium ions out of the cell, making the membrane potential slightly more negative. This is referred to as a 'notch' on the action potential waveform.[12]
There is no obvious phase 1 present in pacemaker cells.
Phase 2
This phase is also known as the "plateau" phase due to the membrane potential remaining almost constant, as the membrane slowly begins to repolarize. This is due to the near balance of charge moving into and out of the cell. During this phase delayed rectifier potassium channels (Iks) allow potassium to leave the cell while L-type calcium channels (activated by the influx of sodium during phase 0) allow the movement of calcium ions into the cell. These calcium ions bind to and open more calcium channels (called ryanodine receptors) located on the sarcoplasmic reticulum within the cell, allowing the flow of calcium out of the SR. These calcium ions are responsible for the contraction of the heart.
Calcium also activates chloride channels called Ito2, which allow Cl− to enter the cell. Increased calcium concentration in the cell also increases activity of the sodium-calcium exchangers, while increased sodium concentration (from the depolarisation of phase 0) increases activity of the sodium-potassium pumps. The movement of all these ions results in the membrane potential remaining relatively constant, with K+ outflux, Cl− influx as well as Na+/K+ pumps contributing to repolarisation and Ca2+ influx as well as Na+/Ca2+ exchangers contributing to depolarisation.[22][12] This phase is responsible for the large duration of the action potential and is important in preventing irregular heartbeat (cardiac arrhythmia).
There is no plateau phase present in pacemaker action potentials.
Phase 3
During phase 3 (the "rapid repolarization" phase) of the action potential, the L-type Ca2+ channels close, while the slow delayed rectifier (IKs) K+ channels remain open as more potassium leak channels open. This ensures a net outward positive current, corresponding to negative change in membrane potential, thus allowing more types of K+ channels to open. These are primarily the rapid delayed rectifier K+ channels (IKr) and the inwardly rectifying K+ current, IK1. This net outward, positive current (equal to loss of positive charge from the cell) causes the cell to repolarize. The delayed rectifier K+ channels close when the membrane potential is restored to about -85 to -90 mV, while IK1 remains conducting throughout phase 4, which helps to set the resting membrane potential[23]
Ionic pumps as discussed above, like the sodium-calcium exchanger and the sodium-potassium pump restore ion concentrations back to balanced states pre-action potential. This means that the intracellular calcium is pumped out, which was responsible for cardiac myocyte contraction. Once this is lost, the contraction stops and the heart muscles relax.
In the sinoatrial node, this phase is also due to the closure of the L-type calcium channels, preventing inward flux of Ca2+ and the opening of the rapid delayed rectifier potassium channels (IKr).[24]
Refractory period
Cardiac cells have two refractory periods, the first from the beginning of phase 0 until part way through phase 3; this is known as the absolute refractory period during which it is impossible for the cell to produce another action potential. This is immediately followed, until the end of phase 3, by a relative refractory period, during which a stronger-than-usual stimulus is required to produce another action potential.[25][26]
These two refractory periods are caused by changes in the states of sodium and potassium channels. The rapid depolarization of the cell, during phase 0, causes the membrane potential to approach sodium's equilibrium potential (i.e. the membrane potential at which sodium is no longer drawn into or out of the cell). As the membrane potential becomes more positive, the sodium channels then close and lock, this is known as the "inactivated" state. During this state the channels cannot be opened regardless of the strength of the excitatory stimulus—this gives rise to the absolute refractory period. The relative refractory period is due to the leaking of potassium ions, which makes the membrane potential more negative (i.e. it is hyperpolarised), this resets the sodium channels; opening the inactivation gate, but still leaving the channel closed. Because some of the voltage-gated sodium ion channels have recovered and the voltage-gated potassium ion channels remain open, it is possible to initiate another action potential if the stimulus is stronger than a stimulus which can fire an action potential when the membrane is at rest.[27]
Gap junctions
Gap junctions allow the action potential to be transferred from one cell to the next (they are said to electrically couple neighbouring cardiac cells). They are made from the connexin family of proteins, that form a pore through which ions (including Na+, Ca2+ and K+) can pass. As potassium is highest within the cell, it is mainly potassium that passes through. This increased potassium in the neighbour cell causes the membrane potential to increase slightly, activating the sodium channels and initiating an action potential in this cell. (A brief chemical gradient driven efflux of Na+ through the connexon at peak depolarization causes the conduction of cell to cell depolarization, not potassium.)[28] These connections allow for the rapid conduction of the action potential throughout the heart and are responsible for allowing all of the cells in the atria to contract together as well as all of the cells in the ventricles.[29] Uncoordinated contraction of heart muscles is the basis for arrhythmia and heart failure.[30]
Channels
| Current (I) | α subunit protein | α subunit gene | Phase / role | |
|---|---|---|---|---|
| Na+ | INa | NaV1.5 | SCN5A[32] | 0 |
| Ca2+ | ICa(L) | CaV1.2 | CACNA1C[33] | 0-2 |
| K+ | Ito1 | KV4.2/4.3 | KCND2/KCND3 | 1, notch |
| K+ | IKs | KV7.1 | KCNQ1 | 2,3 |
| K+ | IKr | KV11.1 (hERG) | KCNH2 | 3 |
| K+ | IK1 | Kir2.1/2.2/2.3 | KCNJ2/KCNJ12/KCNJ4 | 3,4 |
| Na+, Ca2+ | INaCa | 3Na+-1Ca2+-exchanger | NCX1 (SLC8A1) | ion homeostasis |
| Na+, K+ | INaK | 3Na+-2K+-ATPase | ATP1A | ion homeostasis |
| Ca2+ | IpCa | Ca2+-transporting ATPase | ATP1B | ion homeostasis |
Ion channels are proteins that change shape in response to different stimuli to either allow or prevent the movement of specific ions across a membrane. They are said to be selectively permeable. Stimuli, which can either come from outside the cell or from within the cell, can include the binding of a specific molecule to a receptor on the channel (also known as ligand-gated ion channels) or a change in membrane potential around the channel, detected by a sensor (also known as voltage-gated ion channels) and can act to open or close the channel. The pore formed by an ion channel is aqueous (water-filled) and allows the ion to rapidly travel across the membrane.[34] Ion channels can be selective for specific ions, so there are Na+, K+, Ca2+, and Cl− specific channels. They can also be specific for a certain charge of ions (i.e. positive or negative).[35]
Each channel is coded by a set of DNA instructions that tell the cell how to make it. These instructions are known as a gene. Figure 3 shows the important ion channels involved in the cardiac action potential, the current (ions) that flows through the channels, their main protein subunits (building blocks of the channel), some of their controlling genes that code for their structure, and the phases that are active during the cardiac action potential. Some of the most important ion channels involved in the cardiac action potential are described briefly below.
HCN channels
Hyperpolarization-activated cyclic nucleotide-gated channels (HCN channels) are located mainly in pacemaker cells, these channels become active at very negative membrane potentials and allow for the passage of both Na+ and K+ into the cell (which is a movement known as a funny current, If). These poorly selective, cation (positively charged ions) channels conduct more current as the membrane potential becomes more negative (hyperpolarised). The activity of these channels in the SAN cells causes the membrane potential to depolarise slowly and so they are thought to be responsible for the pacemaker potential. Sympathetic nerves directly affect these channels, resulting in an increased heart rate (see below).[36][15]
The fast sodium channel
These sodium channels are voltage-dependent, opening rapidly due to depolarization of the membrane, which usually occurs from neighboring cells, through gap junctions. They allow for a rapid flow of sodium into the cell, depolarizing the membrane completely and initiating an action potential. As the membrane potential increases, these channels then close and lock (become inactive). Due to the rapid influx sodium ions (steep phase 0 in action potential waveform) activation and inactivation of these channels happens almost at exactly the same time. During the inactivation state, Na+ cannot pass through (absolute refractory period). However they begin to recover from inactivation as the membrane potential becomes more negative (relative refractory period).
Potassium channels
The two main types of potassium channels in cardiac cells are inward rectifiers and voltage-gated potassium channels.
Inwardly rectifying potassium channels (Kir) favour the flow of K+ into the cell. This influx of potassium, however, is larger when the membrane potential is more negative than the equilibrium potential for K+ (~-90 mV). As the membrane potential becomes more positive (i.e. during cell stimulation from a neighbouring cell), the flow of potassium into the cell via the Kir decreases. Therefore, Kir is responsible for maintaining the resting membrane potential and initiating the depolarization phase. However, as the membrane potential continues to become more positive, the channel begins to allow the passage of K+ out of the cell. This outward flow of potassium ions at the more positive membrane potentials means that the Kir can also aid the final stages of repolarisation.[37][38]
The voltage-gated potassium channels (Kv) are activated by depolarization. The currents produced by these channels include the transient out potassium current Ito1. This current has two components. Both components activate rapidly, but Ito,fast inactivates more rapidly than Ito, slow. These currents contribute to the early repolarization phase (phase 1) of the action potential.
Another form of voltage-gated potassium channels are the delayed rectifier potassium channels. These channels carry potassium currents which are responsible for the plateau phase of the action potential, and are named based on the speed at which they activate: slowly activating IKs, rapidly activating IKr and ultra-rapidly activating IKur.[39]
Calcium channels
There are two voltage-gated calcium channels within cardiac muscle: L-type calcium channels ('L' for Long-lasting) and T-type calcium channels ('T' for Transient, i.e. short). L-type channels are more common and are most densely populated within the T-tubule membrane of ventricular cells, whereas the T-type channels are found mainly within atrial and pacemaker cells, but still to a lesser degree than L-type channels.
These channels respond to voltage changes across the membrane differently: L-type channels are activated by more positive membrane potentials, take longer to open and remain open longer than T-type channels. This means that the T-type channels contribute more to depolarization (phase 0) whereas L-type channels contribute to the plateau (phase 2).[40]
Conduction system

In the heart's conduction system electrical activity that originates from the sinoatrial node (SAN) is propagated via the His-Purkinje network, the fastest conduction pathway within the heart. The electrical signal travels from the sinoatrial node, which stimulates the atria to contract, to the atrioventricular node (AVN), which slows down conduction of the action potential from the atria to the ventricles. This delay allows the ventricles to fully fill with blood before contraction. The signal then passes down through a bundle of fibres called the bundle of His, located between the ventricles, and then to the Purkinje fibers at the bottom (apex) of the heart, causing ventricular contraction.
In addition to the SAN, the AVN and Purkinje fibres also have pacemaker activity and can therefore spontaneously generate an action potential. However, these cells usually do not depolarize spontaneously, simply because action potential production in the SAN is faster. This means that before the AVN or Purkinje fibres reach the threshold potential for an action potential, they are depolarized by the oncoming impulse from the SAN[41] This is called "overdrive suppression".[42] Pacemaker activity of these cells is vital, as it means that if the SAN were to fail, then the heart could continue to beat, albeit at a lower rate (AVN= 40-60 beats per minute, Purkinje fibres = 20-40 beats per minute). These pacemakers will keep a patient alive until the emergency team arrives.
An example of premature ventricular contraction is the classic athletic heart syndrome. Sustained training of athletes causes a cardiac adaptation where the resting SAN rate is lower (sometimes around 40 beats per minute). This can lead to atrioventricular block, where the signal from the SAN is impaired in its path to the ventricles. This leads to uncoordinated contractions between the atria and ventricles, without the correct delay in between and in severe cases can result in sudden death.[43]
Regulation by the autonomic nervous system
The speed of action potential production in pacemaker cells is affected, but not controlled by the autonomic nervous system.
The sympathetic nervous system (nerves dominant during the body's fight-or-flight response) increase heart rate (positive chronotropy), by decreasing the time to produce an action potential in the SAN. Nerves from the spinal cord release a molecule called noradrenaline, which binds to and activates receptors on the pacemaker cell membrane called β1 adrenoceptors. This activates a protein, called a Gs-protein (s for stimulatory). Activation of this G-protein leads to increased levels of cAMP in the cell (via the cAMP pathway). cAMP binds to the HCN channels (see above), increasing the funny current and therefore increasing the rate of depolarization, during the pacemaker potential. The increased cAMP also increases the opening time of L -type calcium channels, increasing the Ca2+ current through the channel, speeding up phase 0.[44]
The parasympathetic nervous system (nerves dominant while the body is resting and digesting) decreases heart rate (negative chronotropy), by increasing the time taken to produce an action potential in the SAN. A nerve called the vagus nerve, that begins in the brain and travels to the sinoatrial node, releases a molecule called acetylcholine (ACh) which binds to a receptor located on the outside of the pacemaker cell, called an M2 muscarinic receptor. This activates a Gi-protein (I for inhibitory), which is made up of 3 subunits (α, β and γ) which, when activated, separate from the receptor. The β and γ subunits activate a special set of potassium channels, increasing potassium flow out of the cell and decreasing membrane potential, meaning that the pacemaker cells take longer to reach their threshold value.[45] The Gi-protein also inhibits the cAMP pathway therefore reducing the sympathetic effects caused by the spinal nerves.[46]
Clinical significance
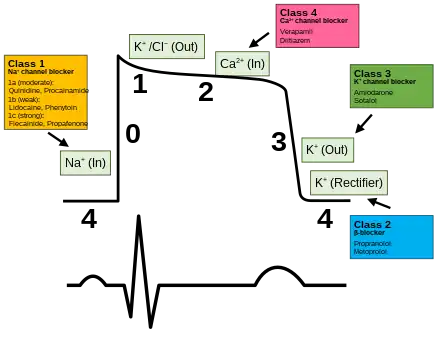
Antiarrhythmic drugs are used to regulate heart rhythms that are too fast. Other drugs used to influence the cardiac action potential include sodium channel blockers, beta blockers, potassium channel blockers, and calcium channel blockers.
References
- ↑ Rudy Y (2008). "Molecular basis of cardiac action potential repolarization". Annals of the New York Academy of Sciences. 1123 (1): 113–8. Bibcode:2008NYASA1123..113R. doi:10.1196/annals.1420.013. PMID 18375583. S2CID 13231624.
- ↑ Zhao, G; Qiu, Y; Zhang, HM; Yang, D (January 2019). "Intercalated discs: cellular adhesion and signaling in heart health and diseases". Heart Failure Reviews. 24 (1): 115–132. doi:10.1007/s10741-018-9743-7. PMID 30288656. S2CID 52919432.
- ↑ Kurtenbach S, Kurtenbach S, Zoidl G (2014). "Gap junction modulation and its implications for heart function". Frontiers in Physiology. 5: 82. doi:10.3389/fphys.2014.00082. PMC 3936571. PMID 24578694.
- ↑ Soltysinska E, Speerschneider T, Winther SV, Thomsen MB (August 2014). "Sinoatrial node dysfunction induces cardiac arrhythmias in diabetic mice". Cardiovascular Diabetology. 13: 122. doi:10.1186/s12933-014-0122-y. PMC 4149194. PMID 25113792.
- ↑ Becker Daniel E (2006). "Fundamentals of Electrocardiography Interpretation". Anesthesia Progress. 53 (2): 53–64. doi:10.2344/0003-3006(2006)53[53:foei]2.0.co;2. PMC 1614214. PMID 16863387.
- ↑ Lote, C. (2012). Principles of Renal Physiology (5th ed.). Springer. p. 150. ISBN 9781461437840.
- 1 2 Santana, Luis F.; Cheng, Edward P.; Lederer, W. Jonathan (2010-12-01). "How does the shape of the cardiac action potential control calcium signaling and contraction in the heart?". Journal of Molecular and Cellular Cardiology. 49 (6): 901–903. doi:10.1016/j.yjmcc.2010.09.005. PMC 3623268. PMID 20850450.
- ↑ Koivumäki, Jussi T.; Korhonen, Topi; Tavi, Pasi (2011-01-01). "Impact of Sarcoplasmic Reticulum Calcium Release on Calcium Dynamics and Action Potential Morphology in Human Atrial Myocytes: A Computational Study". PLOS Computational Biology. 7 (1): e1001067. Bibcode:2011PLSCB...7E1067K. doi:10.1371/journal.pcbi.1001067. PMC 3029229. PMID 21298076.
- ↑ Issa ZF, Miller JM, Zipes DP (2019). "Electrophysiological Mechanisms of Cardiac Arrhythmias: Abnormal Automaticity". In Issa ZF (ed.). Clinical arrhythmology and electrophysiology: a companion to Braunwald's heart disease (Third ed.). Philadelphia, PA: Elsevier. pp. 51–80. doi:10.1016/B978-0-323-52356-1.00003-7. ISBN 978-0-323-52356-1.
- ↑ Antzelevitch C, Burashnikov A (March 2011). "Overview of Basic Mechanisms of Cardiac Arrhythmia". Cardiac Electrophysiology Clinics. 3 (1): 23–45. doi:10.1016/j.ccep.2010.10.012. PMC 3164530. PMID 21892379.
- ↑ Krul S. "Cardiac Arrhythmias - Textbook of Cardiology". www.textbookofcardiology.org. Retrieved 2022-05-17.
- 1 2 3 4 Santana, L.F., Cheng, E.P. and Lederer, J.W. (2010a) 'How does the shape of the cardiac action potential control calcium signaling and contraction in the heart?', 49(6).
- ↑ Morad M., Tung L. (1982). "Ionic events responsible for the cardiac resting and action potential". The American Journal of Cardiology. 49 (3): 584–594. doi:10.1016/s0002-9149(82)80016-7. PMID 6277179.
- ↑ Grunnet M (2010). "Repolarization of the cardiac action potential. Does an increase in repolarization capacity constitute a new anti-arrhythmic principle?". Acta Physiologica. 198: 1–48. doi:10.1111/j.1748-1716.2009.02072.x. PMID 20132149.
- 1 2 DiFrancesco, Dario (2010-02-19). "The role of the funny current in pacemaker activity". Circulation Research. 106 (3): 434–446. doi:10.1161/CIRCRESAHA.109.208041. ISSN 1524-4571. PMID 20167941.
- ↑ Joung B, Chen PS, Lin SF (March 2011). "The role of the calcium and the voltage clocks in sinoatrial node dysfunction". Yonsei Medical Journal. 52 (2): 211–9. doi:10.3349/ymj.2011.52.2.211. PMC 3051220. PMID 21319337.
- ↑ Shih, H T (1994-01-01). "Anatomy of the action potential in the heart". Texas Heart Institute Journal. 21 (1): 30–41. ISSN 0730-2347. PMC 325129. PMID 7514060.
- ↑ Purves et al. 2008, pp. 26–28.
- ↑ Rhoades & Bell 2009, p. 45.
- ↑ Boron, Walter F.; Boulpaep, Emile L. (2012). Medical physiology : a cellular and molecular approach. Boron, Walter F.,, Boulpaep, Emile L. (Updated ed.). Philadelphia, PA. p. 508. ISBN 9781437717532. OCLC 756281854.
{{cite book}}: CS1 maint: location missing publisher (link) - ↑ Sherwood 2012, p. 311.
- ↑ Grunnet M (2010b). "Repolarization of the cardiac action potential. Does an increase in repolarization capacity constitute a new anti-arrhythmic principle?". Acta Physiologica. 198: 1–48. doi:10.1111/j.1748-1716.2009.02072.x. PMID 20132149.
- ↑ Kubo, Y; Adelman, JP; Clapham, DE; Jan, LY; et al. (2005). "International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels". Pharmacol Rev. 57 (4): 509–26. doi:10.1124/pr.57.4.11. PMID 16382105. S2CID 11588492.
- ↑ Clark RB, Mangoni ME, Lueger A, Couette B, Nargeot J, Giles WR (May 2004). "A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells". American Journal of Physiology. Heart and Circulatory Physiology. 286 (5): H1757–66. doi:10.1152/ajpheart.00753.2003. PMID 14693686.
- ↑ Purves et al. 2008, p. 49.
- ↑ Bullock, TH; Orkand, R; Grinnell, A (1977). Introduction to Nervous Systems. New York: W. H. Freeman. p. 151. ISBN 978-0716700302.
- ↑ Sherwood 2008, p. 316.
- ↑ Dubin (2003). Ion Adventure in the Heartland Volume 1. Cover Publishing Company. p. 145. ISBN 978-0-912912-11-0.
- ↑ Goodenough, Daniel A.; Paul, David L. (2009-07-01). "Gap junctions". Cold Spring Harbor Perspectives in Biology. 1 (1): a002576. doi:10.1101/cshperspect.a002576. ISSN 1943-0264. PMC 2742079. PMID 20066080.
- ↑ Severs, Nicholas J. (2002-12-01). "Gap junction remodeling in heart failure". Journal of Cardiac Failure. 8 (6 Suppl): S293–299. doi:10.1054/jcaf.2002.129255. ISSN 1071-9164. PMID 12555135.
- ↑ Sherwood 2008, pp. 248–50.
- ↑ "SCN5A sodium channel, voltage-gated, type V, alpha subunit [Homo sapiens (human)]". National Center for Biotechnology Information.
- ↑ Lacerda, AE; Kim, HS; Ruth, P; Perez-Reyes, E; et al. (August 1991). "Normalization of current kinetics by interaction between the alpha 1 and beta subunits of the skeletal muscle dihydropyridine-sensitive Ca2+ channel". Nature. 352 (6335): 527–30. Bibcode:1991Natur.352..527L. doi:10.1038/352527a0. PMID 1650913. S2CID 4246540.
- ↑ Purves, Dale; Augustine, George J.; Fitzpatrick, David; Katz, Lawrence C.; LaMantia, Anthony-Samuel; McNamara, James O.; Williams, S. Mark (2001-01-01). "The Molecular Structure of Ion Channels". Neuroscience (2nd ed.).
- ↑ Sheng, Morgan. "Ion channels and receptors" (PDF). Retrieved 2013-03-14.
- ↑ Sherwood 2012, pp. 310–1.
- ↑ Hibino, Hiroshi; Inanobe, Atsushi; Furutani, Kazuharu; Murakami, Shingo; Findlay, Ian; Kurachi, Yoshihisa (2010-01-01). "Inwardly rectifying potassium channels: their structure, function, and physiological roles". Physiological Reviews. 90 (1): 291–366. doi:10.1152/physrev.00021.2009. ISSN 1522-1210. PMID 20086079. S2CID 472259.
- ↑ Dhamoon, Amit S.; Jalife, José (2005-03-01). "The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis". Heart Rhythm. 2 (3): 316–324. doi:10.1016/j.hrthm.2004.11.012. ISSN 1547-5271. PMID 15851327.
- ↑ Snyders, D. J. (1999-05-01). "Structure and function of cardiac potassium channels". Cardiovascular Research. 42 (2): 377–390. doi:10.1016/s0008-6363(99)00071-1. ISSN 0008-6363. PMID 10533574.
- ↑ Nargeot, J. (2000-03-31). "A tale of two (Calcium) channels". Circulation Research. 86 (6): 613–615. doi:10.1161/01.res.86.6.613. ISSN 0009-7330. PMID 10746994.
- ↑ Tsien, R. W.; Carpenter, D. O. (1978-06-01). "Ionic mechanisms of pacemaker activity in cardiac Purkinje fibers". Federation Proceedings. 37 (8): 2127–2131. ISSN 0014-9446. PMID 350631.
- ↑ Vassalle, M. (1977). "The relationship among cardiac pacemakers: Overdrive suppression". Circulation Research. 41 (3): 269–77. doi:10.1161/01.res.41.3.269. PMID 330018.
- ↑ Fagard R (2003-12-01). "Athlete's heart". Heart. 89 (12): 1455–61. doi:10.1136/heart.89.12.1455. PMC 1767992. PMID 14617564.
- ↑ DiFrancesco, D.; Tortora, P. (1991-05-09). "Direct activation of cardiac pacemaker channels by intracellular cyclic AMP". Nature. 351 (6322): 145–147. Bibcode:1991Natur.351..145D. doi:10.1038/351145a0. ISSN 0028-0836. PMID 1709448. S2CID 4326191.
- ↑ Osterrieder, W.; Noma, A.; Trautwein, W. (1980-07-01). "On the kinetics of the potassium channel activated by acetylcholine in the S-A node of the rabbit heart". Pflügers Archiv: European Journal of Physiology. 386 (2): 101–109. doi:10.1007/bf00584196. ISSN 0031-6768. PMID 6253873. S2CID 32845421.
- ↑ Demir, Semahat S.; Clark, John W.; Giles, Wayne R. (1999-06-01). "Parasympathetic modulation of sinoatrial node pacemaker activity in rabbit heart: a unifying model". American Journal of Physiology. Heart and Circulatory Physiology. 276 (6): H2221–H2244. doi:10.1152/ajpheart.1999.276.6.H2221. ISSN 0363-6135. PMID 10362707.
Bibliography
- Rudy, Yoram (March 2008). "Molecular Basis of Cardiac Action Potential Repolarization". Annals of the New York Academy of Sciences. 1123 (Control and Regulation of Transport Phenomena in the Cardiac System): 113–8. Bibcode:2008NYASA1123..113R. doi:10.1196/annals.1420.013. PMID 18375583. S2CID 13231624.
- Sherwood, L. (2008). Human Physiology, From Cells to Systems (7th ed.). Cengage Learning. ISBN 9780495391845.
- Sherwood, L. (2012). Human Physiology, From Cells to Systems (8th [revised] ed.). Cengage Learning. ISBN 9781111577438.
- Purves, D; Augustine, GJ; Fitzpatrick, D; Hall, WC; et al. (2008). Neuroscience (4th ed.). Sunderland, MA: Sinauer Associates. ISBN 9780878936977.
- Rhoades, R.; Bell, D.R., eds. (2009). Medical Physiology: Principles for Clinical Medicine. Lippincott Williams & Wilkins. ISBN 9780781768528.
External links
- Interactive animation illustrating the generation of a cardiac action potential
- Interactive mathematical models of cardiac action potential and other generic action potentials