| Voltage gated chloride channel | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 | |||||||||
| Identifiers | |||||||||
| Symbol | Voltage_CLC | ||||||||
| Pfam | PF00654 | ||||||||
| InterPro | IPR014743 | ||||||||
| SCOP2 | 1kpl / SCOPe / SUPFAM | ||||||||
| TCDB | 2.A.49 | ||||||||
| OPM superfamily | 10 | ||||||||
| OPM protein | 1ots | ||||||||
| CDD | cd00400 | ||||||||
| |||||||||
Chloride channels are a superfamily of poorly understood ion channels specific for chloride. These channels may conduct many different ions, but are named for chloride because its concentration in vivo is much higher than other anions.[1] Several families of voltage-gated channels and ligand-gated channels (e.g., the CaCC families) have been characterized in humans.
Voltage-gated chloride channels perform numerous crucial physiological and cellular functions, such as controlling pH, volume homeostasis, transporting organic solutes, regulating cell migration, proliferation, and differentiation. Based on sequence homology the chloride channels can be subdivided into a number of groups.
General functions
Voltage-gated chloride channels are important for setting cell resting membrane potential and maintaining proper cell volume. These channels conduct Cl− or other anions such as HCO−3, I−, SCN−, and NO−3. The structure of these channels are not like other known channels. The chloride channel subunits contain between 1 and 12 transmembrane segments. Some chloride channels are activated only by voltage (i.e., voltage-gated), while others are activated by Ca2+, other extracellular ligands, or pH.[2]
CLC family
The CLC family of chloride channels contains 10 or 12 transmembrane helices. Each protein forms a single pore. It has been shown that some members of this family form homodimers. In terms of primary structure, they are unrelated to known cation channels or other types of anion channels. Three CLC subfamilies are found in animals. CLCN1 is involved in setting and restoring the resting membrane potential of skeletal muscle, while other channels play important parts in solute concentration mechanisms in the kidney.[3] These proteins contain two CBS domains. Chloride channels are also important for maintaining safe ion concentrations within plant cells.[4]
Structure and mechanism
The CLC channel structure has not yet been resolved, however the structure of the CLC exchangers has been resolved by x-ray crystallography. Because the primary structure of the channels and exchangers are so similar, most assumptions about the structure of the channels are based on the structure established for the bacterial exchangers.[5]
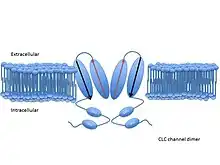
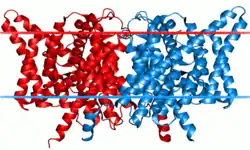
Each channel or exchanger is composed of two similar subunits—a dimer—each subunit containing one pore. The proteins are formed from two copies of the same protein—a homodimer—though scientists have artificially combined subunits from different channels to form heterodimers. Each subunit binds ions independently of the other, meaning conduction or exchange occur independently in each subunit.[3]

Each subunit consists of two related halves oriented in opposite directions, forming an ‘antiparallel’ structure. These halves come together to form the anion pore.[5] The pore has a filter through which chloride and other anions can pass, but lets little else through. These water-filled pores filter anions via three binding sites—Sint, Scen, and Sext—which bind chloride and other anions. The names of these binding sites correspond to their positions within the membrane. Sint is exposed to intracellular fluid, Scen lies inside the membrane or in the center of the filter, and Sext is exposed to extracellular fluid.[4] Each binding site binds different chloride anions simultaneously. In the exchangers, these chloride ions do not interact strongly with one another, due to compensating interactions with the protein. In the channels, the protein does not shield chloride ions at one binding site from the neighboring negatively charged chlorides.[6] Each negative charge exerts a repulsive force on the negative charges next to it. Researchers have suggested that this mutual repulsion contributes to the high rate of conduction through the pore.[5]
CLC transporters shuttle H+ across the membrane. The H+ pathway in CLC transporters utilizes two glutamate residues—one on the extracellular side, Gluex, and one on the intracellular side, Gluin. Gluex also serves to regulate chloride exchange between the protein and extracellular solution. This means that the chloride and the proton share a common pathway on the extracellular side, but diverge on the intracellular side.[6]
CLC channels also have dependence on H+, but for gating rather than Cl− exchange. Instead of utilizing gradients to exchange two Cl− for one H+, the CLC channels transport one H+ while simultaneously transporting millions of anions.[6] This corresponds with one cycle of the slow gate.
Eukaryotic CLC channels also contain cytoplasmic domains. These domains have a pair of CBS motifs, whose function is not fully characterized yet.[5] Though the precise function of these domains is not fully characterized, their importance is illustrated by the pathologies resulting from their mutation. Thomsen's disease, Dent's disease, infantile malignant osteopetrosis, and Bartter's syndrome are all genetic disorders due to such mutations.
At least one role of the cytoplasmic CBS domains regards regulation via adenosine nucleotides. Particular CLC transporters and proteins have modulated activity when bound with ATP, ADP, AMP, or adenosine at the CBS domains. The specific effect is unique to each protein, but the implication is that certain CLC transporters and proteins are sensitive to the metabolic state of the cell.[6]
Selectivity
The Scen acts as the primary selectivity filter for most CLC proteins, allowing the following anions to pass through, from most selected to least: SCN−, Cl−, Br−, NO−
3, I−. Altering a serine residue at the selectivity filter, labeled Sercen, to a different amino acid alters the selectivity.[6]
Gating and kinetics
Gating occurs through two mechanisms: protopore or fast gating and common or slow gating. Common gating involves both protein subunits closing their pores at the same time (cooperation), while protopore gating involves independent opening and closing of each pore.[5] As the names imply, fast gating occur at a much faster rate than slow gating. Precise molecular mechanisms for gating are still being studied.
For the channels, when the slow gate is closed, no ions permeate through the pore. When the slow gate is open, the fast gates open spontaneously and independently of one another. Thus, the protein could have both gates open, or both gates closed, or just one of the two gates open. Single-channel patch-clamp studies demonstrated this biophysical property even before the dual-pore structure of CLC channels had been resolved. Each fast gate opens independently of the other and the ion conductance measured during these studies reflects a binomial distribution.[3]
H+ transport promotes opening of the common gate in CLC channels. For every opening and closing of the common gate, one H+ is transported across the membrane. The common gate is also affected by the bonding of adenosine nucleotides to the intracellular CBS domains. Inhibition or activation of the protein by these domains is specific to each protein.[6]
Function
The CLC channels allow chloride to flow down its electrochemical gradient, when open. These channels are expressed on the cell membrane. CLC channels contribute to the excitability of these membranes as well as transport ions across the membrane.[3]
The CLC exchangers are localized to intracellular components like endosomes or lysosomes and help regulate the pH of their compartments.[3]
Pathology
Bartter's syndrome, which is associated with renal salt wasting and hypokalemic alkalosis, is due to the defective transport of chloride ions and associated ions in the thick ascending loop of Henle. CLCNKB has been implicated.[7]
Another inherited disease that affects the kidney organs is Dent's Disease, characterised by low molecular weight proteinuria and hypercalciuria where mutations in CLCN5 are implicated.[7]
Thomsen disease is associated with dominant mutations and Becker disease with recessive mutations in CLCN1.[7]
Genes
E-ClC family
| CLCA, N-terminal | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | CLCA_N | ||||||||
| Pfam | PF08434 | ||||||||
| InterPro | IPR013642 | ||||||||
| TCDB | 1.A.13 | ||||||||
| |||||||||
Members of Epithelial Chloride Channel (E-ClC) Family (TC# 1.A.13) catalyze bidirectional transport of chloride ions. Mammals have multiple isoforms (at least 6 different gene products plus splice variants) of epithelial chloride channel proteins, catalogued into the Chloride channel accessory (CLCA) family.[8] The first member of this family to be characterized was a respiratory epithelium, Ca2+-regulated, chloride channel protein isolated from bovine tracheal apical membranes.[9] It was biochemically characterized as a 140 kDa complex. The bovine EClC protein has 903 amino acids and four putative transmembrane segments. The purified complex, when reconstituted in a planar lipid bilayer, behaved as an anion-selective channel.[10] It was regulated by Ca2+ via a calmodulin kinase II-dependent mechanism. Distant homologues may be present in plants, ciliates and bacteria, Synechocystis and Escherichia coli, so at least some domains within E-ClC family proteins have an ancient origin.
Genes
CLIC family
| Chloride intracellular ion channel | |
|---|---|
| Identifiers | |
| Symbol | CLIC |
| InterPro | IPR002946 |
| TCDB | 1.A.12 |
The Chloride Intracellular Ion Channel (CLIC) Family (TC# 1.A.12) consists of six conserved proteins in humans (CLIC1, CLIC2, CLIC3, CLIC4, CLIC5, CLIC6). Members exist as both monomeric soluble proteins and integral membrane proteins where they function as chloride-selective ion channels. These proteins are thought to function in the regulation of the membrane potential and in transepithelial ion absorption and secretion in the kidney.[11] They are a member of the glutathione S-transferase (GST) superfamily.
Structure
They possess one or two putative transmembrane α-helical segments (TMSs). The bovine p64 protein is 437 amino acyl residues in length and has the two putative TMSs at positions 223-239 and 367-385. The N- and C-termini are cytoplasmic, and the large central luminal loop may be glycosylated. The human nuclear protein (CLIC1 or NCC27) is much smaller (241 residues) and has only one putative TMS at positions 30-36. It is homologous to the second half of p64.
Structural studies showed that in the soluble form, CLIC proteins adopt a GST fold with an active site exhibiting a conserved glutaredoxin monothiol motif, similar to the omega class GSTs. Al Khamici et al. demonstrated that CLIC proteins have glutaredoxin-like glutathione-dependent oxidoreductase enzymatic activity.[12] CLICs 1, 2 and 4 demonstrate typical glutaredoxin-like activity using 2-hydroxyethyl disulfide as a substrate. This activity may regulate CLIC ion channel function.[12]
Transport reaction
The generalized transport reaction believed to be catalyzed chloride channels is:
- Cl− (cytoplasm) → Cl− (intraorganellar space)
CFTR
CFTR is a chloride channel belonging to the superfamily of ABC transporters. Each channel has two transmembrane domains and two nucleotide binding domains. ATP binding to both nucleotide binding domains causes changes these domains to associate, further causing changes that open up the ion pore. When ATP is hydrolyzed, the nucleotide binding domains dissociate again and the pore closes.[13]
Pathology
Cystic fibrosis is caused by mutations in the CFTR gene on chromosome 7, the most common mutation being deltaF508 (a deletion of a codon coding for phenylalanine, which occupies the 508th amino acid position in the normal CFTR polypeptide). Any of these mutations can prevent the proper folding of the protein and induce its subsequent degradation, resulting in decreased numbers of chloride channels in the body. This causes the buildup of mucus in the body and chronic infections.[13]
Other chloride channels and families
References
- ↑ Jentsch TJ, Stein V, Weinreich F, Zdebik AA (April 2002). "Molecular structure and physiological function of chloride channels". Physiological Reviews. 82 (2): 503–68. doi:10.1152/physrev.00029.2001. PMID 11917096.
- ↑ Suzuki M, Morita T, Iwamoto T (January 2006). "Diversity of Cl(-) channels". Cellular and Molecular Life Sciences. 63 (1): 12–24. doi:10.1007/s00018-005-5336-4. PMC 2792346. PMID 16314923.
- 1 2 3 4 5 Stölting G, Fischer M, Fahlke C (January 2014). "CLC channel function and dysfunction in health and disease". Frontiers in Physiology. 5: 378. doi:10.3389/fphys.2014.00378. PMC 4188032. PMID 25339907.
- ↑ Li WY, Wong FL, Tsai SN, Phang TH, Shao G, Lam HM (June 2006). "Tonoplast-located GmCLC1 and GmNHX1 from soybean enhance NaCl tolerance in transgenic bright yellow (BY)-2 cells". Plant, Cell & Environment. 29 (6): 1122–37. doi:10.1111/j.1365-3040.2005.01487.x. PMID 17080938.
- 1 2 3 4 5 Dutzler R (June 2007). "A structural perspective on ClC channel and transporter function". FEBS Letters. 581 (15): 2839–44. doi:10.1016/j.febslet.2007.04.016. PMID 17452037. S2CID 6365004.
- 1 2 3 4 5 6 Accardi A, Picollo A (August 2010). "CLC channels and transporters: proteins with borderline personalities". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1798 (8): 1457–64. doi:10.1016/j.bbamem.2010.02.022. PMC 2885512. PMID 20188062.
- 1 2 3 Planells-Cases R, Jentsch TJ (March 2009). "Chloride channelopathies" (PDF). Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1792 (3): 173–89. doi:10.1016/j.bbadis.2009.02.002. PMID 19708126.
- ↑ Evans SR, Thoreson WB, Beck CL (October 2004). "Molecular and functional analyses of two new calcium-activated chloride channel family members from mouse eye and intestine". The Journal of Biological Chemistry. 279 (40): 41792–800. doi:10.1074/jbc.M408354200. PMC 1383427. PMID 15284223.
- ↑ Agnel M, Vermat T, Culouscou JM (July 1999). "Identification of three novel members of the calcium-dependent chloride channel (CaCC) family predominantly expressed in the digestive tract and trachea". FEBS Letters. 455 (3): 295–301. doi:10.1016/s0014-5793(99)00891-1. PMID 10437792. S2CID 82094058.
- ↑ Brunetti E, Filice C (June 1996). "Percutaneous aspiration in the treatment of hydatid liver cysts". Gut. 38 (6): 936. doi:10.1136/gut.38.6.936. PMC 1383206. PMID 8984037.
- ↑ Singh H, Ashley RH (2007-02-01). "CLIC4 (p64H1) and its putative transmembrane domain form poorly selective, redox-regulated ion channels". Molecular Membrane Biology. 24 (1): 41–52. doi:10.1080/09687860600927907. PMID 17453412. S2CID 9986497.
- 1 2 Al Khamici H, Brown LJ, Hossain KR, Hudson AL, Sinclair-Burton AA, Ng JP, Daniel EL, Hare JE, Cornell BA, Curmi PM, Davey MW, Valenzuela SM (2015-01-01). "Members of the chloride intracellular ion channel protein family demonstrate glutaredoxin-like enzymatic activity". PLOS ONE. 10 (1): e115699. Bibcode:2015PLoSO..10k5699A. doi:10.1371/journal.pone.0115699. PMC 4291220. PMID 25581026.
- 1 2 Gadsby DC, Vergani P, Csanády L (March 2006). "The ABC protein turned chloride channel whose failure causes cystic fibrosis". Nature. 440 (7083): 477–83. Bibcode:2006Natur.440..477G. doi:10.1038/nature04712. PMC 2720541. PMID 16554808.
Further reading
- Schmidt-Rose T, Jentsch TJ (August 1997). "Reconstitution of functional voltage-gated chloride channels from complementary fragments of CLC-1". The Journal of Biological Chemistry. 272 (33): 20515–21. doi:10.1074/jbc.272.33.20515. PMID 9252364.
- Zhang J, George AL, Griggs RC, Fouad GT, Roberts J, Kwieciński H, Connolly AM, Ptácek LJ (October 1996). "Mutations in the human skeletal muscle chloride channel gene (CLCN1) associated with dominant and recessive myotonia congenita". Neurology. 47 (4): 993–8. doi:10.1212/wnl.47.4.993. PMID 8857733. S2CID 45062016.
- Mindell JA, Maduke M (2001). "ClC chloride channels". Genome Biology. 2 (2): REVIEWS3003. doi:10.1186/gb-2001-2-2-reviews3003. PMC 138906. PMID 11182894.
- Singh H (May 2010). "Two decades with dimorphic Chloride Intracellular Channels (CLICs)". FEBS Letters. 584 (10): 2112–21. doi:10.1016/j.febslet.2010.03.013. PMID 20226783. S2CID 21056278.
External links
- Chloride+channels at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- UMich Orientation of Proteins in Membranes families/superfamily-10 - CLC chloride channels
As of this edit, this article uses content from "1.A.13 The Epithelial Chloride Channel (E-ClC) Family", which is licensed in a way that permits reuse under the Creative Commons Attribution-ShareAlike 3.0 Unported License, but not under the GFDL. All relevant terms must be followed. As of this edit, this article uses content from "1.A.12 The Intracellular Chloride Channel (CLIC) Family", which is licensed in a way that permits reuse under the Creative Commons Attribution-ShareAlike 3.0 Unported License, but not under the GFDL. All relevant terms must be followed.