Epoxidation with dioxiranes refers to the synthesis of epoxides from alkenes using three-membered cyclic peroxides, also known as dioxiranes.[1]
Dioxiranes are three-membered cyclic peroxides containing a weak oxygen-oxygen bond. Although they are able to effect oxidations of heteroatom functionality and even carbon-hydrogen bonds,[2] they are most widely used as epoxidizing agents of alkenes. Dioxiranes are electrophilic oxidants that react more quickly with electron-rich than electron-poor double bonds; however, both classes of substrates can be epoxidized within a reasonable time frame. Dioxiranes may be prepared and isolated or generated in situ from ketones and potassium peroxymonosulfate (Oxone). In situ preparations may be catalytic in ketone, and if the ketone is chiral, enantioselective epoxidation takes place. The functional group compatibility of dioxiranes is limited somewhat, as side oxidations of amines and sulfides are rapid. Nonetheless, protocols for dioxirane oxidations are entirely metal free. The most common dioxiranes employed for synthesis are dimethyl dioxirane (DMD) and methyl(trifluoromethyl)dioxirane (TFD).

Mechanism and stereochemistry
The mechanism of epoxidation with dioxiranes most likely involves concerted oxygen transfer through a spiro transition state.[3] As oxygen transfer occurs, the plane of the oxirane is perpendicular to and bisects the plane of the alkene pi system. The configuration of the alkene is maintained in the product, ruling out long-lived radical intermediates. In addition, the spiro transition state has been used to explain the sense of selectivity in enantioselective epoxidations with chiral ketones.[4]

Stereoselective variants
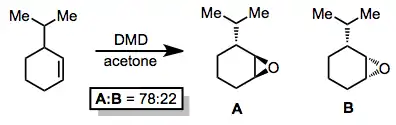
Diastereoselective epoxidation may be achieved through the use of alkene starting materials with diastereotopic faces. When racemic 3-isopropylcyclohexene was subjected to DMD oxidation, the trans epoxide, which resulted from attack on the less hindered face of the double bond, was the major product.[5]
Enantioselective epoxidation using dioxiranes exploits one of two strategies: (1) oxidation by DMD of a chiral metal catalyst followed by epoxidation, or (2) epoxidation by chiral dioxiranes, which are generated in situ from a catalytic amount of ketone and a stoichiometric amount of a terminal oxidant).[4] Mn-salen complexes have been used with success to accomplish the first strategy.[6]

Many of the best ketones for the second strategy are derived from carbohydrates. For instance, Shi's catalyst 1 is derived from fructose, and epoxidizes both di- and trisubstituted alkenes with high enantioselectivity.[4]

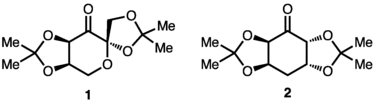
Scope and limitations
Dioxiranes may either be prepared in advance or generated in situ for epoxidation reactions. In most cases, a two-phase system must be set up for in situ epoxidations, as KHSO5 is not soluble in organic solvents. Thus, substrates or products sensitive to hydrolysis will not survive in situ epoxidations.[7] This section describes epoxidation conditions for alkenes with electron-donating or -withdrawing substituents, both of which may be epoxidized with dioxiranes in either the stoichiometric or catalytic mode.
Although dioxiranes are highly electrophilic, they epoxidize both electron-rich and electron-poor alkenes in good yield (although the latter react much more slowly). Electron-poor epoxide products also exhibit enhanced hydrolytic stability, meaning that they can often survive in situ conditions. Epoxidations of electron-rich double bonds have yielded intermediates of Rubottom oxidation. Upon hydrolysis, these siloxyepoxides yield α-hydroxyketones.[8]

Electron-poor double bonds take much longer to epoxidize. Heating may be used to encourage oxidation, although the reaction temperature should never exceed 50 °C, to avoid decomposition of the dioxirane.[9]

Alkenes bound to both electron-withdrawing and -donating groups tend to behave like the former, requiring long oxidation times and occasionally some heating. Like electron-poor epoxides, epoxide products from this class of substrates are often stable with respect to hydrolysis.[10]

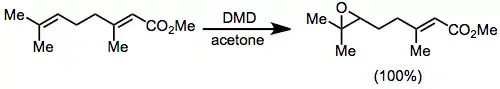
In substrates containing multiple double bonds, the most electron-rich double bond can usually be selectively epoxidized.[11]
Epoxidations employing aqueous Oxone and a catalytic amount of ketone are convenient if a specialized dioxirane must be used (as in asymmetric applications) or if isolation of the dioxirane is inconvenient. Hydrolytic decomposition of the epoxidation product may be used to good advantage.[12]

Synthetic applications
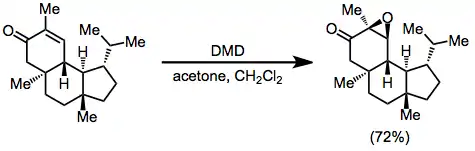
Diastereoselective DMD epoxidation of a chiral unsaturated ketone was applied to the synthesis of verrucosan-2β-ol.[13]
Enantioselective dioxirane epoxidation is critical in a synthetic sequence leading to an analogue of glabrescol. The sequence produced the glabrescol analogue in 31% overall yield in only two steps.[14]

Comparison with other methods
Dioxirane epoxidation is highly versatile, and compares favorably to related peracid oxidations in many respects. Peracids generate acidic byproducts, meaning that acid-labile substrates and products must be avoided.[15] Dioxirane epoxidations using isolated oxidant can be carried out under neutral conditions without the need for aqueous buffering. However, catalytic dioxirane oxidations do require water and are not suitable for hydrolytically unstable substrates.
Some methods are well-suited to the oxidation of electron-rich or electron-poor double bonds, but few are as effective for both classes of substrate as dioxiranes. Weitz-Scheffer conditions (NaOCl, H2O2/KOH, tBuO2H/KOH) work well for oxidations of electron-poor double bonds,[16] and sulfonyl-substituted oxaziridines are effective for electron-rich double bonds.[17]
Metal-based oxidants are often more efficient than dioxirane oxidations in the catalytic mode; however, environmentally unfriendly byproducts are typically generated. In the realm of asymmetric methods, both the Sharpless epoxidation[18] and Jacobsen epoxidation[19] surpass asymmetric dioxirane oxidations in enantioselectivity. Additionally, enzymatic epoxidations are more enantioselective than dioxirane-based methods; however, operational difficulties and low yields are sometimes associated with enzymatic oxidations[20]
Experimental conditions
Dioxiranes are generated by combining the ketone precursor with a buffered aqueous solution of KHSO5. The volatile dioxiranes DMD and TFD are isolated via distillation of the crude reaction mixture. Baeyer-Villiger oxidation may compete with dioxirane formation. Once isolated, dioxiranes are kept in solutions of the corresponding ketones and dried with molecular sieves. Air-free technique is unnecessary unless the substrate or product is air-sensitive or hydrolytically labile, and most oxidations are carried out in the open air in Erlenmeyer flasks.
Oxidations with in situ generated dioxiranes are more convenient than isolation methods, provided the substrate is stable towards hydrolysis. Reactions can either be carried out in truly biphasic media with mechanical stirring, or in a homogeneous medium derived from water and a miscible organic solvent, such as acetonitrile. Asymmetric epoxidations are commonly carried out under the latter conditions. Some ketone catalysts are more persistent under slightly basic homogeneous conditions.
References
- ↑ Adam, W.; Saha-Moller, C.; Zhao, C.-G. Org. React. 2003, 61, 219. doi:10.1002/0471264180.or061.02
- ↑ Adam, W.; Zhao, C.-G.; Jakka, K. Org. React. 2007, 69, 1.
- ↑ Houk, K. N.; Liu, J.; DeMello, N. C.; Condroski, K. R. J. Am. Chem. Soc. 1997, 119, 10147.
- 1 2 3 Wang, Z.-X.; Tu, Y.; Frohn, M.; Zhang, J.-R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224.
- ↑ Adam, W.; Mitchell, C. M.; Saha-Möller, C. R. Eur. J. Org. Chem. 1999, 785.
- ↑ Lévai, A.; Adam, W.; Fell, R. T.; Gessner, R.; Patonay, T.; Simon, A.; Tóth, G. Tetrahedron 1998, 54, 13105.
- ↑ Denmark, S. E.; Wu, Z. J. Org. Chem. 1998, 63, 2810.
- ↑ Adam, W.; Hadjiarapoglou, L.; Wang, X. Tetrahedron Lett. 1989, 30, 6497.
- ↑ Adam, W.; Hadjiarapoglou, L.; Nestler, B. Tetrahedron Lett. 1990, 31, 331.
- ↑ Yang, D.; Wong, M.-K.; Yip, Y.-C. J. Org. Chem. 1995, 60, 3887.
- ↑ Messeguer, A.; Fusco, C.; Curci, R. Tetrahedron 1993, 49, 6299.
- ↑ Denmark, S. E.; Forbes, D. C.; Hays, D. S.; DePue, J. S.; Wilde, R. G. J. Org. Chem. 1995, 60, 1391.
- ↑ Piers, E.; Boulet, S. L. Tetrahedron Lett. 1997, 38, 8815.
- ↑ Xiong, Z.; Corey, E. J. J. Am. Chem. Soc. 2000, 122, 4831.
- ↑ Dryuk, V. G. Russ. Chem. Rev. 1985, 54, 986.
- ↑ Patai, S.; Rappoport, Z. In The Chemistry of Alkenes; Patai, S., Ed.; Wiley: New York, 1964, Vol. 1, pp. 512–517.
- ↑ Davis, F. A.; Sheppard, A. C.; Chen, B.-C.; Haque, M. S. J. Am. Chem. Soc. 1990, 112, 6679.
- ↑ Katsuki, T.; Martin, V. S. Org. React. 1996, 48, 1.
- ↑ Jacobsen, E. N. In Comprehensive Organometallic Chemistry II; Abel, E. W.; Stone, F. G. A.; Wilkinson, G.; Hegedus, L. S., Eds.; Pergamon: New York, 1995, Vol. 12, Chapter 11.1, pp. 1097–1135.
- ↑ Adam, W.; Lazarus, M.; Saha-Möller, C. R.; Weichold, O.; Hoch, U.; Häring, D.; Schreier P. In Advances in Biochemical Engineering/Biotechnology; Faber, K., Ed.; Springer Verlag: Heidelberg, 1999, Vol. 63, pp. 73–108.