Flow cytometry bioinformatics is the application of bioinformatics to flow cytometry data, which involves storing, retrieving, organizing and analyzing flow cytometry data using extensive computational resources and tools. Flow cytometry bioinformatics requires extensive use of and contributes to the development of techniques from computational statistics and machine learning. Flow cytometry and related methods allow the quantification of multiple independent biomarkers on large numbers of single cells. The rapid growth in the multidimensionality and throughput of flow cytometry data, particularly in the 2000s, has led to the creation of a variety of computational analysis methods, data standards, and public databases for the sharing of results.
Computational methods exist to assist in the preprocessing of flow cytometry data, identifying cell populations within it, matching those cell populations across samples, and performing diagnosis and discovery using the results of previous steps. For preprocessing, this includes compensating for spectral overlap, transforming data onto scales conducive to visualization and analysis, assessing data for quality, and normalizing data across samples and experiments. For population identification, tools are available to aid traditional manual identification of populations in two-dimensional scatter plots (gating), to use dimensionality reduction to aid gating, and to find populations automatically in higher-dimensional space in a variety of ways. It is also possible to characterize data in more comprehensive ways, such as the density-guided binary space partitioning technique known as probability binning, or by combinatorial gating. Finally, diagnosis using flow cytometry data can be aided by supervised learning techniques, and discovery of new cell types of biological importance by high-throughput statistical methods, as part of pipelines incorporating all of the aforementioned methods.
Open standards, data and software are also key parts of flow cytometry bioinformatics. Data standards include the widely adopted Flow Cytometry Standard (FCS) defining how data from cytometers should be stored, but also several new standards under development by the International Society for Advancement of Cytometry (ISAC) to aid in storing more detailed information about experimental design and analytical steps. Open data is slowly growing with the opening of the CytoBank database in 2010, and FlowRepository in 2012, both of which allow users to freely distribute their data, and the latter of which has been recommended as the preferred repository for MIFlowCyt-compliant data by ISAC. Open software is most widely available in the form of a suite of Bioconductor packages, but is also available for web execution on the GenePattern platform.
Data collection
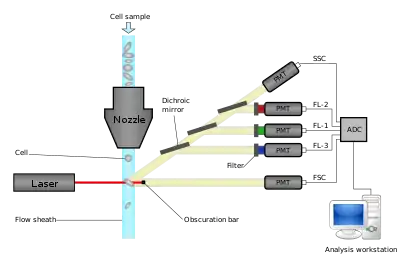
Flow cytometers operate by hydrodynamically focusing suspended cells so that they separate from each other within a fluid stream. The stream is interrogated by one or more lasers, and the resulting fluorescent and scattered light is detected by photomultipliers. By using optical filters, particular fluorophores on or within the cells can be quantified by peaks in their emission spectra. These may be endogenous fluorophores such as chlorophyll or transgenic green fluorescent protein, or they may be artificial fluorophores covalently bonded to detection molecules such as antibodies for detecting proteins, or hybridization probes for detecting DNA or RNA.
The ability to quantify these has led to flow cytometry being used in a wide range of applications, including but not limited to:
- Monitoring of CD4 count in HIV[1]
- Diagnosis of various cancers[2][3]
- Analysis of aquatic microbiomes[4]
- Sperm sorting[5]
- Measuring telomere length[6]
Until the early 2000s, flow cytometry could only measure a few fluorescent markers at a time. Through the late 1990s into the mid-2000s, however, rapid development of new fluorophores resulted in modern instruments capable of quantifying up to 18 markers per cell.[7] More recently, the new technology of mass cytometry replaces fluorophores with rare-earth elements detected by time of flight mass spectrometry, achieving the ability to measure the expression of 34 or more markers.[8] At the same time, microfluidic qPCR methods are providing a flow cytometry-like method of quantifying 48 or more RNA molecules per cell.[9] The rapid increase in the dimensionality of flow cytometry data, coupled with the development of high-throughput robotic platforms capable of assaying hundreds to thousands of samples automatically have created a need for improved computational analysis methods.[7]
Data
Flow cytometry data is in the form of a large matrix of intensities over M wavelengths by N events. Most events will be a particular cell, although some may be doublets (pairs of cells which pass the laser closely together). For each event, the measured fluorescence intensity over a particular wavelength range is recorded.
The measured fluorescence intensity indicates the amount of that fluorophore in the cell, which indicates the amount that has bound to detector molecules such as antibodies. Therefore, fluorescence intensity can be considered a proxy for the amount of detector molecules present on the cell. A simplified, if not strictly accurate, way of considering flow cytometry data is as a matrix of M measurements times N cells where each element corresponds to the amounts of molecules.
Steps in computational flow cytometry data analysis
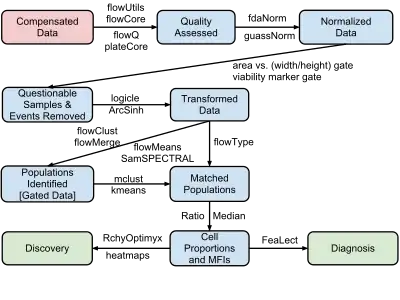
The process of moving from primary FCM data to disease diagnosis and biomarker discovery involves four major steps:
- Data pre-processing (including compensation, transformation and normalization)
- Cell population identification (a.k.a. gating)
- Cell population matching for cross sample comparison
- Relating cell populations to external variables (diagnosis and discovery)
Saving of the steps taken in a particular flow cytometry workflow is supported by some flow cytometry software, and is important for the reproducibility of flow cytometry experiments. However, saved workspace files are rarely interchangeable between software.[10] An attempt to solve this problem is the development of the Gating-ML XML-based data standard (discussed in more detail under the standards section), which is slowly being adopted in both commercial and open source flow cytometry software.[11] The CytoML R package is also filling the gap by importing/exporting the Gating-ML that is compatible with FlowJo, CytoBank and FACS Diva softwares.
Data pre-processing
Prior to analysis, flow cytometry data must typically undergo pre-processing to remove artifacts and poor quality data, and to be transformed onto an optimal scale for identifying cell populations of interest. Below are various steps in a typical flow cytometry preprocessing pipeline.
Compensation
When more than one fluorochrome is used with the same laser, their emission spectra frequently overlap. Each particular fluorochrome is typically measured using a bandpass optical filter set to a narrow band at or near the fluorochrome's emission intensity peak. The result is that the reading for any given fluorochrome is actually the sum of that fluorochrome's peak emission intensity, and the intensity of all other fluorochromes' spectra where they overlap with that frequency band. This overlap is termed spillover, and the process of removing spillover from flow cytometry data is called compensation.[12]
Compensation is typically accomplished by running a series of representative samples each stained for only one fluorochrome, to give measurements of the contribution of each fluorochrome to each channel.[12] The total signal to remove from each channel can be computed by solving a system of linear equations based on this data to produce a spillover matrix, which when inverted and multiplied with the raw data from the cytometer produces the compensated data.[12][13] The processes of computing the spillover matrix, or applying a precomputed spillover matrix to compensate flow cytometry data, are standard features of flow cytometry software.[14]
Transformation
Cell populations detected by flow cytometry are often described as having approximately log-normal expression.[15] As such, they have traditionally been transformed to a logarithmic scale. In early cytometers, this was often accomplished even before data acquisition by use of a log amplifier. On modern instruments, data is usually stored in linear form, and transformed digitally prior to analysis.
However, compensated flow cytometry data frequently contains negative values due to compensation, and cell populations do occur which have low means and normal distributions.[16] Logarithmic transformations cannot properly handle negative values, and poorly display normally distributed cell types.[16][17] Alternative transformations which address this issue include the log-linear hybrid transformations Logicle[16][18] and Hyperlog,[19] as well as the hyperbolic arcsine and the Box–Cox.[20]
A comparison of commonly used transformations concluded that the biexponential and Box–Cox transformations, when optimally parameterized, provided the clearest visualization and least variance of cell populations across samples.[17] However, a later comparison of the flowTrans package used in that comparison indicated that it did not parameterize the Logicle transformation in a manner consistent with other implementations, potentially calling those results into question.[21]
Quality control
Particularly in newer, high-throughput experiments, there is a need for visualization methods to help detect technical errors in individual samples. One approach is to visualize summary statistics, such as the empirical distribution functions of single dimensions of technical or biological replicates to ensure they are the similar.[22] For more rigor, the Kolmogorov–Smirnov test can be used to determine if individual samples deviate from the norm.[22] The Grubbs's test for outliers may be used to detect samples deviating from the group.
A method for quality control in higher-dimensional space is to use probability binning with bins fit to the whole data set pooled together.[23] Then the standard deviation of the number of cells falling in the bins within each sample can be taken as a measure of multidimensional similarity, with samples that are closer to the norm having a smaller standard deviation.[23] With this method, higher standard deviation can indicate outliers, although this is a relative measure as the absolute value depends partly on the number of bins.
With all of these methods, the cross-sample variation is being measured. However, this is the combination of technical variations introduced by the instruments and handling, and actual biological information that is desired to be measured. Disambiguating the technical and the biological contributions to between-sample variation can be a difficult to impossible task.[24]
Normalization
Particularly in multi-centre studies, technical variation can make biologically equivalent populations of cells difficult to match across samples. Normalization methods to remove technical variance, frequently derived from image registration techniques, are thus a critical step in many flow cytometry analyses. Single-marker normalization can be performed using landmark registration, in which peaks in a kernel density estimate of each sample are identified and aligned across samples.[24]
Identifying cell populations

The complexity of raw flow cytometry data (dozens of measurements for thousands to millions of cells) makes answering questions directly using statistical tests or supervised learning difficult. Thus, a critical step in the analysis of flow cytometric data is to reduce this complexity to something more tractable while establishing common features across samples. This usually involves identifying multidimensional regions that contain functionally and phenotypically homogeneous groups of cells.[27] This is a form of cluster analysis. There are a range of methods by which this can be achieved, detailed below.
Gating
The data generated by flow-cytometers can be plotted in one or two dimensions to produce a histogram or scatter plot. The regions on these plots can be sequentially separated, based on fluorescence intensity, by creating a series of subset extractions, termed "gates". These gates can be produced using software, e.g. FlowJo,[28] FCS Express,[29] WinMDI,[30] CytoPaint (aka Paint-A-Gate),[31] VenturiOne, Cellcion, CellQuest Pro, Cytospec,[32] Kaluza.[33] or flowCore.
In datasets with a low number of dimensions and limited cross-sample technical and biological variability (e.g., clinical laboratories), manual analysis of specific cell populations can produce effective and reproducible results. However, exploratory analysis of a large number of cell populations in a high-dimensional dataset is not feasible.[34] In addition, manual analysis in less controlled settings (e.g., cross-laboratory studies) can increase the overall error rate of the study.[35] In one study, several computational gating algorithms performed better than manual analysis in the presence of some variation.[26] However, despite the considerable advances in computational analysis, manual gating remains the main solution for the identification of specific rare cell populations that are not well-separated from other cell types.
Gating guided by dimension reduction
The number of scatter plots that need to be investigated increases with the square of the number of markers measured (or faster since some markers need to be investigated several times for each group of cells to resolve high-dimensional differences between cell types that appear to be similar in most markers).[36] To address this issue, principal component analysis has been used to summarize the high-dimensional datasets using a combination of markers that maximizes the variance of all data points.[37] However, PCA is a linear method and is not able to preserve complex and non-linear relationships. More recently, two dimensional minimum spanning tree layouts have been used to guide the manual gating process. Density-based down-sampling and clustering was used to better represent rare populations and control the time and memory complexity of the minimum spanning tree construction process.[38] More sophisticated dimension reduction algorithms are yet to be investigated.[39]
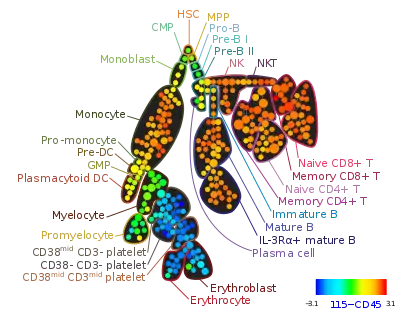
Automated gating
Developing computational tools for identification of cell populations has been an area of active research only since 2008. Many individual clustering approaches have recently been developed, including model-based algorithms (e.g., flowClust[41] and FLAME[42]), density based algorithms (e.g. FLOCK[43] and SWIFT, graph-based approaches (e.g. SamSPECTRAL[44]) and most recently, hybrids of several approaches (flowMeans[45] and flowPeaks[46]). These algorithms are different in terms of memory and time complexity, their software requirements, their ability to automatically determine the required number of cell populations, and their sensitivity and specificity. The FlowCAP (Flow Cytometry: Critical Assessment of Population Identification Methods) project, with active participation from most academic groups with research efforts in the area, is providing a way to objectively cross-compare state-of-the-art automated analysis approaches.[26] Other surveys have also compared automated gating tools on several datasets.[47][48][49][50]
Probability binning methods
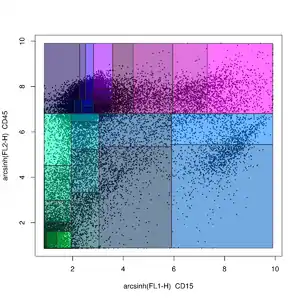
Probability binning is a non-gating analysis method in which flow cytometry data is split into quantiles on a univariate basis.[51] The locations of the quantiles can then be used to test for differences between samples (in the variables not being split) using the chi-squared test.[51]
This was later extended into multiple dimensions in the form of frequency difference gating, a binary space partitioning technique where data is iteratively partitioned along the median.[52] These partitions (or bins) are fit to a control sample. Then the proportion of cells falling within each bin in test samples can be compared to the control sample by the chi squared test.
Finally, cytometric fingerprinting uses a variant of frequency difference gating to set bins and measure for a series of samples how many cells fall within each bin.[23] These bins can be used as gates and used for subsequent analysis similarly to automated gating methods.
Combinatorial gating
High-dimensional clustering algorithms are often unable to identify rare cell types that are not well separated from other major populations. Matching these small cell populations across multiple samples is even more challenging. In manual analysis, prior biological knowledge (e.g., biological controls) provides guidance to reasonably identify these populations. However, integrating this information into the exploratory clustering process (e.g., as in semi-supervised learning) has not been successful.
An alternative to high-dimensional clustering is to identify cell populations using one marker at a time and then combine them to produce higher-dimensional clusters. This functionality was first implemented in FlowJo.[28] The flowType algorithm builds on this framework by allowing the exclusion of the markers.[53] This enables the development of statistical tools (e.g. RchyOptimyx) that can investigate the importance of each marker and exclude high-dimensional redundancies.[54]
Diagnosis and discovery

After identification of the cell population of interest, a cross sample analysis can be performed to identify phenotypical or functional variations that are correlated with an external variable (e.g., a clinical outcome). These studies can be partitioned into two main groups:
Diagnosis
In these studies, the goal usually is to diagnose a disease (or a sub-class of a disease) using variations in one or more cell populations. For example, one can use multidimensional clustering to identify a set of clusters, match them across all samples, and then use supervised learning to construct a classifier for prediction of the classes of interest (e.g., this approach can be used to improve the accuracy of the classification of specific lymphoma subtypes[55]). Alternatively, all the cells from the entire cohort can be pooled into a single multidimensional space for clustering before classification.[56] This approach is particularly suitable for datasets with a high amount of biological variation (in which cross-sample matching is challenging) but requires technical variations to be carefully controlled.[57]
Discovery
In a discovery setting, the goal is to identify and describe cell populations correlated with an external variable (as opposed to the diagnosis setting in which the goal is to combine the predictive power of multiple cell types to maximize the accuracy of the results). Similar to the diagnosis use-case, cluster matching in high-dimensional space can be used for exploratory analysis but the descriptive power of this approach is very limited, as it is hard to characterize and visualize a cell population in a high-dimensional space without first reducing the dimensionality.[56][58] Finally, combinatorial gating approaches have been particularly successful in exploratory analysis of FCM data. Simplified Presentation of Incredibly Complex Evaluations (SPICE) is a software package that can use the gating functionality of FlowJo to statistically evaluate a wide range of different cell populations and visualize those that are correlated with the external outcome. flowType and RchyOptimyx (as discussed above) expand this technique by adding the ability of exploring the impact of independent markers on the overall correlation with the external outcome. This enables the removal of unnecessary markers and provides a simple visualization of all identified cell types. In a recent analysis of a large (n=466) cohort of HIV+ patients, this pipeline identified three correlates of protection against HIV, only one of which had been previously identified through extensive manual analysis of the same dataset.[53]
Data formats and interchange
Flow Cytometry Standard
Flow Cytometry Standard (FCS) was developed in 1984 to allow recording and sharing of flow cytometry data.[59] Since then, FCS became the standard file format supported by all flow cytometry software and hardware vendors. The FCS specification has traditionally been developed and maintained by the International Society for Advancement of Cytometry (ISAC).[60] Over the years, updates were incorporated to adapt to technological advancements in both flow cytometry and computing technologies with FCS 2.0 introduced in 1990,[61] FCS 3.0 in 1997,[62] and the most current specification FCS 3.1 in 2010.[63] FCS used to be the only widely adopted file format in flow cytometry. Recently, additional standard file formats have been developed by ISAC.
netCDF
ISAC is considering replacing FCS with a flow cytometry specific version of the Network Common Data Form (netCDF) file format.[64] netCDF is a set of freely available software libraries and machine independent data formats that support the creation, access, and sharing of array-oriented scientific data. In 2008, ISAC drafted the first version of netCDF conventions for storage of raw flow cytometry data.[65]
Archival Cytometry Standard (ACS)
The Archival Cytometry Standard (ACS) is being developed to bundle data with different components describing cytometry experiments.[66] It captures relations among data, metadata, analysis files and other components, and includes support for audit trails, versioning and digital signatures. The ACS container is based on the ZIP file format with an XML-based table of contents specifying relations among files in the container. The XML Signature W3C Recommendation has been adopted to allow for digital signatures of components within the ACS container. An initial draft of ACS has been designed in 2007 and finalized in 2010. Since then, ACS support has been introduced in several software tools including FlowJo and Cytobank.
Gating-ML
The lack of gating interoperability has traditionally been a bottleneck preventing reproducibility of flow cytometry data analysis and the usage of multiple analytical tools. To address this shortcoming, ISAC developed Gating-ML, an XML-based mechanism to formally describe gates and related data (scale) transformations.[10] The draft recommendation version of Gating-ML was approved by ISAC in 2008 and it is partially supported by tools like FlowJo, the flowUtils, CytoML libraries in R/BioConductor, and FlowRepository.[66] It supports rectangular gates, polygon gates, convex polytopes, ellipsoids, decision trees and Boolean collections of any of the other types of gates. In addition, it includes dozens of built in public transformations that have been shown to potentially useful for display or analysis of cytometry data. In 2013, Gating-ML version 2.0 was approved by ISAC's Data Standards Task Force as a Recommendation. This new version offers slightly less flexibility in terms of the power of gating description; however, it is also significantly easier to implement in software tools.[11]
Classification Results (CLR)
The Classification Results (CLR) File Format[67] has been developed to exchange the results of manual gating and algorithmic classification approaches in a standard way in order to be able to report and process the classification. CLR is based in the commonly supported CSV file format with columns corresponding to different classes and cell values containing the probability of an event being a member of a particular class. These are captured as values between 0 and 1. Simplicity of the format and its compatibility with common spreadsheet tools have been the major requirements driving the design of the specification. Although it was originally designed for the field of flow cytometry, it is applicable in any domain that needs to capture either fuzzy or unambiguous classifications of virtually any kinds of objects.
Public data and software
As in other bioinformatics fields, development of new methods has primarily taken the form of free open source software, and several databases have been created for depositing open data.
AutoGate
AutoGate[68] performs compensation, gating, preview of clusters, exhaustive projection pursuit (EPP), multi-dimension scaling and phenogram, produces a visual dendogram to express HiD readiness. It is free to researchers and clinicians at academic, government, and non-profit institutions.
Bioconductor
The Bioconductor project is a repository of free open source software, mostly written in the R programming language.[69] As of July 2013, Bioconductor contained 21 software packages for processing flow cytometry data.[70] These packages cover most of the range of functionality described earlier in this article.
GenePattern
GenePattern is a predominantly genomic analysis platform with over 200 tools for analysis of gene expression, proteomics, and other data. A web-based interface provides easy access to these tools and allows the creation of automated analysis pipelines enabling reproducible research. Recently, a GenePattern Flow Cytometry Suite has been developed in order to bring advanced flow cytometry data analysis tools to experimentalists without programmatic skills. It contains close to 40 open source GenePattern flow cytometry modules covering methods from basic processing of flow cytometry standard (i.e., FCS) files to advanced algorithms for automated identification of cell populations, normalization and quality assessment. Internally, most of these modules leverage functionality developed in BioConductor.
Much of the functionality of the Bioconductor packages for flow cytometry analysis has been packaged up for use with the GenePattern[71] workflow system, in the form of the GenePattern Flow Cytometry Suite.[72]
FACSanadu
FACSanadu[73] is an open source portable application for visualization and analysis of FCS data. Unlike Bioconductor, it is an interactive program aimed at non-programmers for routine analysis. It supports standard FCS files as well as COPAS profile data.
hema.to
hema.to is a web service for the classification of flow cytometry data of patients suspected to have lymphoma.[74] The artificial intelligence within the tool uses a deep convolutional neural network to recognize patterns of distinct subtypes. All data and code is open access.[75] It processes raw data, which makes gating unnecessary. For best performance on new data, fine tuning by knowledge transfer is required.[76]
Public databases
The Minimum Information about a Flow Cytometry Experiment (MIFlowCyt), requires that any flow cytometry data used in a publication be available, although this does not include a requirement that it be deposited in a public database.[77] Thus, although the journals Cytometry Part A and B, as well as all journals from the Nature Publishing Group require MIFlowCyt compliance, there is still relatively little publicly available flow cytometry data. Some efforts have been made towards creating public databases, however.
Firstly, CytoBank, which is a complete web-based flow cytometry data storage and analysis platform, has been made available to the public in a limited form.[78] Using the CytoBank code base, FlowRepository was developed in 2012 with the support of ISAC to be a public repository of flow cytometry data.[79] FlowRepository facilitates MIFlowCyt compliance,[80] and as of July 2013 contained 65 public data sets.[81]
Datasets
In 2012, the flow cytometry community has started to release a set of publicly available datasets. A subset of these datasets representing the existing data analysis challenges is described below. For comparison against manual gating, the FlowCAP-I project has released five datasets, manually gated by human analysts, and two of them gated by eight independent analysts.[26] The FlowCAP-II project included three datasets for binary classification and also reported several algorithms that were able to classify these samples perfectly. FlowCAP-III included two larger datasets for comparison against manual gates as well as one more challenging sample classification dataset. As of March 2013, public release of FlowCAP-III was still in progress.[82] The datasets used in FlowCAP-I, II, and III either have a low number of subjects or parameters. However, recently several more complex clinical datasets have been released including a dataset of 466 HIV-infected subjects, which provides both 14 parameter assays and sufficient clinical information for survival analysis.[54][83][84][85]
Another class of datasets are higher-dimensional mass cytometry assays. A representative of this class of datasets is a study which includes analysis of two bone marrow samples using more than 30 surface or intracellular markers under a wide range of different stimulations.[8] The raw data for this dataset is publicly available as described in the manuscript, and manual analyses of the surface markers are available upon request from the authors.
Open problems
Despite rapid development in the field of flow cytometry bioinformatics, several problems remain to be addressed.
Variability across flow cytometry experiments arises from biological variation among samples, technical variations across instruments used, as well as methods of analysis. In 2010, a group of researchers from Stanford University and the National Institutes of Health pointed out that while technical variation can be ameliorated by standardizing sample handling, instrument setup and choice of reagents, solving variation in analysis methods will require similar standardization and computational automation of gating methods.[86] They further opined that centralization of both data and analysis could aid in decreasing variability between experiments and in comparing results.[86]
This was echoed by another group of Pacific Biosciences and Stanford University researchers, who suggested that cloud computing could enable centralized, standardized, high-throughput analysis of flow cytometry experiments.[87] They also emphasised that ongoing development and adoption of standard data formats could continue to aid in reducing variability across experiments.[87] They also proposed that new methods will be needed to model and summarize results of high-throughput analysis in ways that can be interpreted by biologists,[87] as well as ways of integrating large-scale flow cytometry data with other high-throughput biological information, such as gene expression, genetic variation, metabolite levels and disease states.[87]
See also
References
![]() This article was adapted from the following source under a CC BY 4.0 license (2013) (reviewer reports):
Kieran O'Neill; Nima Aghaeepour; Josef Spidlen; Ryan Brinkman (5 December 2013). "Flow cytometry bioinformatics". PLOS Computational Biology. 9 (12): e1003365. doi:10.1371/JOURNAL.PCBI.1003365. ISSN 1553-734X. PMC 3867282. PMID 24363631. Wikidata Q21045422.
This article was adapted from the following source under a CC BY 4.0 license (2013) (reviewer reports):
Kieran O'Neill; Nima Aghaeepour; Josef Spidlen; Ryan Brinkman (5 December 2013). "Flow cytometry bioinformatics". PLOS Computational Biology. 9 (12): e1003365. doi:10.1371/JOURNAL.PCBI.1003365. ISSN 1553-734X. PMC 3867282. PMID 24363631. Wikidata Q21045422.{{cite journal}}: CS1 maint: unflagged free DOI (link)
- ↑ Brando, B.; Barnett, D.; Janossy, G.; Mandy, F.; Autran, B.; Rothe, G.; Scarpati, B.; d'Avanzo, G.; d'Hautcourt, J. L.; Lenkei, R.; Schmitz, G.; Kunkl, A.; Chianese, R.; Papa, S.; Gratama, J. W. (2000). "Cytofluorometric methods for assessing absolute numbers of cell subsets in blood". Cytometry. 42 (6): 327–346. doi:10.1002/1097-0320(20001215)42:6<327::AID-CYTO1000>3.0.CO;2-F. PMID 11135287. S2CID 13492197.
- ↑ Ferreira-Facio, C. S.; Milito, C.; Botafogo, V.; Fontana, M.; Thiago, L. S.; Oliveira, E.; Da Rocha-Filho, A. S.; Werneck, F.; Forny, D. N.; Dekermacher, S.; De Azambuja, A. P.; Ferman, S. E.; De Faria, P. A. N. S.; Land, M. G. P.; Orfao, A.; Costa, E. S. (2013). Aziz, Syed A (ed.). "Contribution of Multiparameter Flow Cytometry Immunophenotyping to the Diagnostic Screening and Classification of Pediatric Cancer". PLOS ONE. 8 (3): e55534. Bibcode:2013PLoSO...855534F. doi:10.1371/journal.pone.0055534. PMC 3589426. PMID 23472067.
- ↑ Wu, D.; Wood, B. L.; Fromm, J. R. (2013). "Flow Cytometry for Non-Hodgkin and Classical Hodgkin Lymphoma". Lymphoma. Methods in Molecular Biology. Vol. 971. pp. 27–47. doi:10.1007/978-1-62703-269-8_2. ISBN 978-1-62703-268-1. PMID 23296956.
- ↑ Wang, Y.; Hammes, F.; De Roy, K.; Verstraete, W.; Boon, N. (2010). "Past, present and future applications of flow cytometry in aquatic microbiology". Trends in Biotechnology. 28 (8): 416–424. doi:10.1016/j.tibtech.2010.04.006. PMID 20541271.
- ↑ Johnson, L. A.; Flook, J. P.; Look, M. V.; Pinkel, D. (1987). "Flow sorting of X and Y chromosome-bearing spermatozoa into two populations". Gamete Research. 16 (1): 1–9. doi:10.1002/mrd.1120160102. PMID 3506896.
- ↑ Baerlocher, G. M.; Vulto, I.; De Jong, G.; Lansdorp, P. M. (2006). "Flow cytometry and FISH to measure the average length of telomeres (flow FISH)". Nature Protocols. 1 (5): 2365–2376. doi:10.1038/nprot.2006.263. PMID 17406480. S2CID 20463557.
- 1 2 Chattopadhyay, P. K.; Hogerkorp, C. M.; Roederer, M. (2008). "A chromatic explosion: The development and future of multiparameter flow cytometry". Immunology. 125 (4): 441–449. doi:10.1111/j.1365-2567.2008.02989.x. PMC 2612557. PMID 19137647.
- 1 2 Behbehani, G. K.; Bendall, S. C.; Clutter, M. R.; Fantl, W. J.; Nolan, G. P. (2012). "Single-cell mass cytometry adapted to measurements of the cell cycle". Cytometry Part A. 81A (7): 552–566. doi:10.1002/cyto.a.22075. PMC 3667754. PMID 22693166.
- ↑ White, A. K.; Vaninsberghe, M.; Petriv, O. I.; Hamidi, M.; Sikorski, D.; Marra, M. A.; Piret, J.; Aparicio, S.; Hansen, C. L. (2011). "High-throughput microfluidic single-cell RT-qPCR". Proceedings of the National Academy of Sciences. 108 (34): 13999–14004. Bibcode:2011PNAS..10813999W. doi:10.1073/pnas.1019446108. PMC 3161570. PMID 21808033.
- 1 2 Spidlen, J.; Leif, R. C.; Moore, W.; Roederer, M.; Brinkman, R. R.; Brinkman, R. R. (2008). "Gating-ML: XML-based gating descriptions in flow cytometry". Cytometry Part A. 73A (12): 1151–1157. doi:10.1002/cyto.a.20637. PMC 2585156. PMID 18773465.
- 1 2 Gating-ML 2.0 (PDF) (Report). International Society for the Advancement of Cytometry. 2013.
- 1 2 3 Roederer, M. (2002). J. Paul Robinson (ed.). Compensation in Flow Cytometry. Current Protocols in Cytometry. Vol. 22. pp. 1.14.1–1.14.20. doi:10.1002/0471142956.cy0114s22. ISBN 978-0471142959. PMID 18770762. S2CID 7256386.
- ↑ Bagwell, C. B.; Adams, E. G. (1993). "Fluorescence spectral overlap compensation for any number of flow cytometry parameters". Annals of the New York Academy of Sciences. 677 (1): 167–184. Bibcode:1993NYASA.677..167B. doi:10.1111/j.1749-6632.1993.tb38775.x. PMID 8494206. S2CID 26763280.
- ↑ Hahne, F.; Lemeur, N.; Brinkman, R. R.; Ellis, B.; Haaland, P.; Sarkar, D.; Spidlen, J.; Strain, E.; Gentleman, R. (2009). "FlowCore: A Bioconductor package for high throughput flow cytometry". BMC Bioinformatics. 10: 106. doi:10.1186/1471-2105-10-106. PMC 2684747. PMID 19358741.
- ↑ Shapiro, Howard M. (2003). Practical flow cytometry. New York: Wiley-Liss. p. 235. ISBN 978-0-471-41125-3.
- 1 2 3 Parks DR, Roederer M, Moore WA (2006). "A new "Logicle" display method avoids deceptive effects of logarithmic scaling for low signals and compensated data". Cytometry Part A. 69 (6): 541–51. doi:10.1002/cyto.a.20258. PMID 16604519. S2CID 8012792.
- 1 2 Finak, G.; Perez, J. M.; Weng, A.; Gottardo, R. (2010). "Optimizing transformations for automated, high throughput analysis of flow cytometry data". BMC Bioinformatics. 11: 546. doi:10.1186/1471-2105-11-546. PMC 3243046. PMID 21050468.
- ↑ Moore, W. A.; Parks, D. R. (2012). "Update for the logicle data scale including operational code implementations". Cytometry Part A. 81A (4): 273–277. doi:10.1002/cyto.a.22030. PMC 4761345. PMID 22411901.
- ↑ Bagwell, C. B. (2005). "Hyperlog?A flexible log-like transform for negative, zero, and positive valued data". Cytometry Part A. 64A (1): 34–42. doi:10.1002/cyto.a.20114. PMID 15700280. S2CID 13705174.
- ↑ Lo, K.; Brinkman, R. R.; Gottardo, R. (2008). "Automated gating of flow cytometry data via robust model-based clustering". Cytometry Part A. 73A (4): 321–332. doi:10.1002/cyto.a.20531. PMID 18307272. S2CID 2943705.
- ↑ Qian, Y.; Liu, Y.; Campbell, J.; Thomson, E.; Kong, Y. M.; Scheuermann, R. H. (2012). "FCSTrans: An open source software system for FCS file conversion and data transformation". Cytometry Part A. 81A (5): 353–356. doi:10.1002/cyto.a.22037. PMC 3932304. PMID 22431383.
- 1 2 Le Meur, N.; Rossini, A.; Gasparetto, M.; Smith, C.; Brinkman, R. R.; Gentleman, R. (2007). "Data quality assessment of ungated flow cytometry data in high throughput experiments". Cytometry Part A. 71A (6): 393–403. doi:10.1002/cyto.a.20396. PMC 2768034. PMID 17366638.
- 1 2 3 Rogers, W. T.; Moser, A. R.; Holyst, H. A.; Bantly, A.; Mohler, E. R.; Scangas, G.; Moore, J. S. (2008). "Cytometric fingerprinting: Quantitative characterization of multivariate distributions". Cytometry Part A. 73A (5): 430–441. doi:10.1002/cyto.a.20545. PMID 18383310. S2CID 23555926.
- 1 2 Hahne, F.; Khodabakhshi, A. H.; Bashashati, A.; Wong, C. J.; Gascoyne, R. D.; Weng, A. P.; Seyfert-Margolis, V.; Bourcier, K.; Asare, A.; Lumley, T.; Gentleman, R.; Brinkman, R. R. (2009). "Per-channel basis normalization methods for flow cytometry data". Cytometry Part A. 77 (2): 121–131. doi:10.1002/cyto.a.20823. PMC 3648208. PMID 19899135.
- ↑ "CLUE package". Retrieved 2013-02-15.
- 1 2 3 4 Aghaeepour, N.; Finak, G.; Flowcap, D.; Dream, A. H.; Hoos, P.; Mosmann, G.; Brinkman, J.; Gottardo, I.; Scheuermann, S. A.; Bramson, J.; Eaves, C.; Weng, A. P.; Iii, E. S. F.; Ho, K.; Kollmann, T.; Rogers, W.; De Rosa, S.; Dalal, B.; Azad, A.; Pothen, A.; Brandes, A.; Bretschneider, H.; Bruggner, R.; Finck, R.; Jia, R.; Zimmerman, N.; Linderman, M.; Dill, D.; Nolan, G.; Chan, C. (2013). "Critical assessment of automated flow cytometry data analysis techniques". Nature Methods. 10 (3): 228–238. doi:10.1038/nmeth.2365. PMC 3906045. PMID 23396282.
- ↑ Lugli, E.; Roederer, M.; Cossarizza, A. (2010). "Data analysis in flow cytometry: The future just started". Cytometry Part A. 77A (7): 705–713. doi:10.1002/cyto.a.20901. PMC 2909632. PMID 20583274.
- 1 2 "FlowJo". Archived from the original on 2013-05-03. Retrieved 2013-04-05.
- ↑ "FCS Express". Retrieved 2013-04-03.
- ↑ "TSRI Cytometry Software Page". Archived from the original on 1996-11-19. Retrieved 2009-09-03.
- ↑ "CytoPaint Classic". Retrieved 2013-04-05.
- ↑ "PUCL Cytometry Software Page". Retrieved 2011-07-07.
- ↑ "Beckman Coulter". Retrieved 2013-02-10.
- ↑ Bendall, S. C.; Nolan, G. P. (2012). "From single cells to deep phenotypes in cancer". Nature Biotechnology. 30 (7): 639–647. doi:10.1038/nbt.2283. PMID 22781693. S2CID 163651.
- ↑ Maecker, H. T.; Rinfret, A.; d'Souza, P.; Darden, J.; Roig, E.; Landry, C.; Hayes, P.; Birungi, J.; Anzala, O.; Garcia, M.; Harari, A.; Frank, I.; Baydo, R.; Baker, M.; Holbrook, J.; Ottinger, J.; Lamoreaux, L.; Epling, C. L.; Sinclair, E.; Suni, M. A.; Punt, K.; Calarota, S.; El-Bahi, S.; Alter, G.; Maila, H.; Kuta, E.; Cox, J.; Gray, C.; Altfeld, M.; Nougarede, N. (2005). "Standardization of cytokine flow cytometry assays". BMC Immunology. 6: 13. doi:10.1186/1471-2172-6-13. PMC 1184077. PMID 15978127.
- ↑ Virgo, P. F.; Gibbs, G. J. (2011). "Flow cytometry in clinical pathology". Annals of Clinical Biochemistry. 49 (Pt 1): 17–28. doi:10.1258/acb.2011.011128. PMID 22028426.
- ↑ Costa, E. S.; Pedreira, C. E.; Barrena, S.; Lecrevisse, Q.; Flores, J.; Quijano, S.; Almeida, J.; Del Carmen García-Macias, M.; Bottcher, S.; Van Dongen, J. J. M.; Orfao, A. (2010). "Automated pattern-guided principal component analysis vs expert-based immunophenotypic classification of B-cell chronic lymphoproliferative disorders: A step forward in the standardization of clinical immunophenotyping". Leukemia. 24 (11): 1927–1933. doi:10.1038/leu.2010.160. PMC 3035971. PMID 20844562.
- ↑ Qiu, P.; Simonds, E. F.; Bendall, S. C.; Gibbs Jr, K. D.; Bruggner, R. V.; Linderman, M. D.; Sachs, K.; Nolan, G. P.; Plevritis, S. K. (2011). "Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE". Nature Biotechnology. 29 (10): 886–891. doi:10.1038/nbt.1991. PMC 3196363. PMID 21964415.
- ↑ "Matlab Toolbox for Dimensionality Reduction". Retrieved 2013-02-10.
- ↑ Bendall, S. C.; Simonds, E. F.; Qiu, P.; Amir, E. -A. D.; Krutzik, P. O.; Finck, R.; Bruggner, R. V.; Melamed, R.; Trejo, A.; Ornatsky, O. I.; Balderas, R. S.; Plevritis, S. K.; Sachs, K.; Pe'Er, D.; Tanner, S. D.; Nolan, G. P. (2011). "Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum". Science. 332 (6030): 687–696. Bibcode:2011Sci...332..687B. doi:10.1126/science.1198704. PMC 3273988. PMID 21551058.
- ↑ Lo, K.; Hahne, F.; Brinkman, R. R.; Gottardo, R. (2009). "FlowClust: A Bioconductor package for automated gating of flow cytometry data". BMC Bioinformatics. 10: 145. doi:10.1186/1471-2105-10-145. PMC 2701419. PMID 19442304.
- ↑ Pyne, S.; Hu, X.; Wang, K.; Rossin, E.; Lin, T. -I.; Maier, L. M.; Baecher-Allan, C.; McLachlan, G. J.; Tamayo, P.; Hafler, D. A.; De Jager, P. L.; Mesirov, J. P. (2009). "Automated high-dimensional flow cytometric data analysis". Proceedings of the National Academy of Sciences. 106 (21): 8519–8524. Bibcode:2009PNAS..106.8519P. doi:10.1073/pnas.0903028106. PMC 2682540. PMID 19443687.
- ↑ Qian, Y.; Wei, C.; Eun-Hyung Lee, F.; Campbell, J.; Halliley, J.; Lee, J. A.; Cai, J.; Kong, Y. M.; Sadat, E.; Thomson, E.; Dunn, P.; Seegmiller, A. C.; Karandikar, N. J.; Tipton, C. M.; Mosmann, T.; Sanz, I. A.; Scheuermann, R. H. (2010). "Elucidation of seventeen human peripheral blood B-cell subsets and quantification of the tetanus response using a density-based method for the automated identification of cell populations in multidimensional flow cytometry data". Cytometry Part B. 78B (Suppl 1): S69–S82. doi:10.1002/cyto.b.20554. PMC 3084630. PMID 20839340.
- ↑ Zare, H.; Shooshtari, P.; Gupta, A.; Brinkman, R. R. (2010). "Data reduction for spectral clustering to analyze high throughput flow cytometry data". BMC Bioinformatics. 11: 403. doi:10.1186/1471-2105-11-403. PMC 2923634. PMID 20667133.
- ↑ Aghaeepour, N.; Nikolic, R.; Hoos, H. H.; Brinkman, R. R. (2011). "Rapid cell population identification in flow cytometry data". Cytometry Part A. 79A (1): 6–13. doi:10.1002/cyto.a.21007. PMC 3137288. PMID 21182178.
- ↑ Ge, Y.; Sealfon, S. C. (2012). "FlowPeaks: A fast unsupervised clustering for flow cytometry data via K-means and density peak finding". Bioinformatics. 28 (15): 2052–2058. doi:10.1093/bioinformatics/bts300. PMC 3400953. PMID 22595209.
- ↑ Weber, Lukas; Robinson, Mark. "Comparison of Clustering Methods for High-Dimensional Single-Cell Flow and Mass Cytometry Data". bioRxiv 10.1101/047613.
- ↑ Chester, C (2015). "Algorithmic tools for mining high-dimensional cytometry data". Journal of Immunology. 195 (3): 773–779. doi:10.4049/jimmunol.1500633. PMC 4507289. PMID 26188071.
- ↑ Diggins, KE (2015). "Methods for discovery and characterization of cell subsets in high dimensional mass cytometry data". Methods. 82: 55–63. doi:10.1016/j.ymeth.2015.05.008. PMC 4468028. PMID 25979346.
- ↑ Wiwie, C (2015). "Comparing the performance of biomedical clustering methods". Nature Methods. 12 (11): 1033–1038. doi:10.1038/nmeth.3583. PMID 26389570. S2CID 8960399.
- 1 2 Roederer, M.; Treister, A.; Moore, W.; Herzenberg, L. A. (2001). "Probability binning comparison: A metric for quantitating univariate distribution differences". Cytometry. 45 (1): 37–46. doi:10.1002/1097-0320(20010901)45:1<37::AID-CYTO1142>3.0.CO;2-E. PMID 11598945.
- ↑ Roederer, M.; Hardy, R. R. (2001). "Frequency difference gating: A multivariate method for identifying subsets that differ between samples". Cytometry. 45 (1): 56–64. doi:10.1002/1097-0320(20010901)45:1<56::AID-CYTO1144>3.0.CO;2-9. PMID 11598947.
- 1 2 3 Aghaeepour, N.; Chattopadhyay, P. K.; Ganesan, A.; O'Neill, K.; Zare, H.; Jalali, A.; Hoos, H. H.; Roederer, M.; Brinkman, R. R. (2012). "Early immunologic correlates of HIV protection can be identified from computational analysis of complex multivariate T-cell flow cytometry assays". Bioinformatics. 28 (7): 1009–1016. doi:10.1093/bioinformatics/bts082. PMC 3315712. PMID 22383736.
- 1 2 3 Aghaeepour, N.; Jalali, A.; O'Neill, K.; Chattopadhyay, P. K.; Roederer, M.; Hoos, H. H.; Brinkman, R. R. (2012). "RchyOptimyx: Cellular hierarchy optimization for flow cytometry". Cytometry Part A. 81A (12): 1022–1030. doi:10.1002/cyto.a.22209. PMC 3726344. PMID 23044634.
- ↑ Zare, H.; Bashashati, A.; Kridel, R.; Aghaeepour, N.; Haffari, G.; Connors, J. M.; Gascoyne, R. D.; Gupta, A.; Brinkman, R. R.; Weng, A. P. (2011). "Automated Analysis of Multidimensional Flow Cytometry Data Improves Diagnostic Accuracy Between Mantle Cell Lymphoma and Small Lymphocytic Lymphoma". American Journal of Clinical Pathology. 137 (1): 75–85. doi:10.1309/AJCPMMLQ67YOMGEW. PMC 4090220. PMID 22180480.
- 1 2 Qiu, P. (2012). Ma’Ayan, Avi (ed.). "Inferring Phenotypic Properties from Single-Cell Characteristics". PLOS ONE. 7 (5): e37038. Bibcode:2012PLoSO...737038Q. doi:10.1371/journal.pone.0037038. PMC 3360688. PMID 22662133.
- ↑ Bodenmiller, B.; Zunder, E. R.; Finck, R.; Chen, T. J.; Savig, E. S.; Bruggner, R. V.; Simonds, E. F.; Bendall, S. C.; Sachs, K.; Krutzik, P. O.; Nolan, G. P. (2012). "Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators". Nature Biotechnology. 30 (9): 858–867. doi:10.1038/nbt.2317. PMC 3627543. PMID 22902532.
- ↑ Bashashati, A.; Johnson, N. A.; Khodabakhshi, A. H.; Whiteside, M. D.; Zare, H.; Scott, D. W.; Lo, K.; Gottardo, R.; Brinkman, F. S. L.; Connors, J. M.; Slack, G. W.; Gascoyne, R. D.; Weng, A. P.; Brinkman, R. R. (2012). "B Cells with High Side Scatter Parameter by Flow Cytometry Correlate with Inferior Survival in Diffuse Large B-Cell Lymphoma". American Journal of Clinical Pathology. 137 (5): 805–814. doi:10.1309/AJCPGR8BG4JDVOWR. PMC 3718075. PMID 22523221.
- ↑ Murphy, R. F.; Chused, T. M. (1984). "A proposal for a flow cytometric data file standard". Cytometry. 5 (5): 553–555. doi:10.1002/cyto.990050521. PMID 6489069.
- ↑ "International Society for Advancement of Cytometry". Retrieved 5 March 2013.
- ↑ Dean, P. N.; Bagwell, C. B.; Lindmo, T.; Murphy, R. F.; Salzman, G. C. (1990). "Introduction to flow cytometry data file standard". Cytometry. 11 (3): 321–322. doi:10.1002/cyto.990110302. PMID 2340768.
- ↑ Seamer, L. C.; Bagwell, C. B.; Barden, L.; Redelman, D.; Salzman, G. C.; Wood, J. C. S.; Murphy, R. F. (1997). "Proposed new data file standard for flow cytometry, version FCS 3.0". Cytometry. 28 (2): 118–122. doi:10.1002/(SICI)1097-0320(19970601)28:2<118::AID-CYTO3>3.0.CO;2-B. PMID 9181300.
- ↑ Spidlen, J.; Moore, W.; Parks, D.; Goldberg, M.; Bray, C.; Bierre, P.; Gorombey, P.; Hyun, B.; Hubbard, M.; Lange, S.; Lefebvre, R.; Leif, R.; Novo, D.; Ostruszka, L.; Treister, A.; Wood, J.; Murphy, R. F.; Roederer, M.; Sudar, D.; Zigon, R.; Brinkman, R. R. (2009). "Data File Standard for Flow Cytometry, version FCS 3.1". Cytometry Part A. 77 (1): 97–100. doi:10.1002/cyto.a.20825. PMC 2892967. PMID 19937951.
- ↑ Robert C. Leif, Josef Spidlen, Ryan R. Brinkman (2009). Farkas, Daniel L; Nicolau, Dan V; Leif, Robert C (eds.). "Cytometry Standards Continuum" (PDF). SPIE Proceedings. Imaging, Manipulation, and Analysis of Biomolecules, Cells, and Tissues VI. 6859: 17. Bibcode:2008SPIE.6859E..17L. CiteSeerX 10.1.1.397.3647. doi:10.1117/12.762514. S2CID 62650477.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ International Society for the Advancement of Cytometry (2008). Analytical Cytometry Standard NetCDF Conventions for List Mode Binary Data File Component
- 1 2 Spidlen, J.; Shooshtari, P.; Kollmann, T. R.; Brinkman, R. R. (2011). "Flow cytometry data standards". BMC Research Notes. 4: 50. doi:10.1186/1756-0500-4-50. PMC 3060130. PMID 21385382.
- ↑ Classification Results File Format (PDF) (Report). International Society for the Advancement of Cytometry. 2012.
- ↑ "CytoGenie - Home page for AutoGate software". CytoGenie.org. Herzenberg Laboratory at Stanford University. Retrieved 14 January 2020.
- ↑ Gentleman, R. C.; Carey, V. J.; Bates, D. M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; Hornik, K.; Hothorn, T.; Huber, W.; Iacus, S.; Irizarry, R.; Leisch, F.; Li, C.; Maechler, M.; Rossini, A. J.; Sawitzki, G.; Smith, C.; Smyth, G.; Tierney, L.; Yang, J. Y.; Zhang, J. (2004). "Bioconductor: Open software development for computational biology and bioinformatics". Genome Biology. 5 (10): R80. doi:10.1186/gb-2004-5-10-r80. PMC 545600. PMID 15461798.
- ↑ Bioconductor. "BioConductor FlowCytometry view". Retrieved 11 July 2013.
- ↑ Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J. P. (2006). "GenePattern 2.0". Nature Genetics. 38 (5): 500–501. doi:10.1038/ng0506-500. PMID 16642009. S2CID 5503897.
- ↑ "GenePattern Flow Cytometry Suite". Archived from the original on 29 January 2013. Retrieved 14 February 2013.
- ↑ "FACSanadu - Free and easy to use FCS analysis software".
- ↑ Zhao, Max; Mallesh, Nanditha; Höllein, Alexander; Schabath, Richard; Haferlach, Claudia; Haferlach, Torsten; Elsner, Franz; Lüling, Hannes; Krawitz, Peter; Kern, Wolfgang (2020). "Hematologist-Level Classification of Mature B-Cell Neoplasm Using Deep Learning on Multiparameter Flow Cytometry Data". Cytometry Part A. 97 (10): 1073–1080. doi:10.1002/cyto.a.24159. ISSN 1552-4930. PMID 32519455. S2CID 219563125.
- ↑ Zhao, Max Xiaohang; Mallesh, Nanditha (2021-06-16). "Replication Data for: Knowledge transfer to enhance the performance of deep learning models for automated classification of B-cell neoplasms". Harvard Dataverse. doi:10.7910/DVN/CQHHEH.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Mallesh, Nanditha; Zhao, Max; Meintker, Lisa; Höllein, Alexander; Elsner, Franz; Lüling, Hannes; Haferlach, Torsten; Kern, Wolfgang; Westermann, Jörg; Brossart, Peter; Krause, Stefan W. (September 2021). "Knowledge transfer to enhance the performance of deep learning models for automated classification of B cell neoplasms". Patterns. 2 (10): 100351. doi:10.1016/j.patter.2021.100351. ISSN 2666-3899. PMC 8515009. PMID 34693376.
- ↑ Lee, J. A.; Spidlen, J.; Boyce, K.; Cai, J.; Crosbie, N.; Dalphin, M.; Furlong, J.; Gasparetto, M.; Goldberg, M.; Goralczyk, E. M.; Hyun, B.; Jansen, K.; Kollmann, T.; Kong, M.; Leif, R.; McWeeney, S.; Moloshok, T. D.; Moore, W.; Nolan, G.; Nolan, J.; Nikolich-Zugich, J.; Parrish, D.; Purcell, B.; Qian, Y.; Selvaraj, B.; Smith, C.; Tchuvatkina, O.; Wertheimer, A.; Wilkinson, P.; Wilson, C. (2008). "MIFlowCyt: The minimum information about a flow cytometry experiment". Cytometry Part A. 73A (10): 926–930. doi:10.1002/cyto.a.20623. PMC 2773297. PMID 18752282.
- ↑ Kotecha, N.; Krutzik, P. O.; Irish, J. M. (2010). J. Paul Robinson (ed.). Web-Based Analysis and Publication of Flow Cytometry Experiments. Current Protocols in Cytometry. Vol. Chapter 10. pp. 10.17.1–10.17.24. doi:10.1002/0471142956.cy1017s53. ISBN 978-0471142959. PMC 4208272. PMID 20578106.
- ↑ Spidlen, J.; Breuer, K.; Rosenberg, C.; Kotecha, N.; Brinkman, R. R. (2012). "FlowRepository: A resource of annotated flow cytometry datasets associated with peer-reviewed publications". Cytometry Part A. 81A (9): 727–731. doi:10.1002/cyto.a.22106. PMID 22887982. S2CID 6498066.
- ↑ Spidlen, J.; Breuer, K.; Brinkman, R. (2012). "Preparing a Minimum Information about a Flow Cytometry Experiment (MIFlowCyt) Compliant Manuscript Using the International Society for Advancement of Cytometry (ISAC) FCS File Repository (FlowRepository.org)". In J. Paul Robinson (ed.). Preparing a Minimum Information about a Flow Cytometry Experiment (MIFlow Cyt) Compliant Manuscript Using the International Society for Advancement of Cytometry (ISAC) FCS File Repository (Flow Repository.org). Current Protocols in Cytometry. Vol. Chapter 10. pp. Unit Un10.18. doi:10.1002/0471142956.cy1018s61. ISBN 978-0471142959. PMID 22752950. S2CID 24921940.
- ↑ "FlowRepository".
- ↑ "FlowCAP - Flow Cytometry: Critical Assessment of Population Identification Methods". Retrieved 15 March 2013.
- ↑ "IDCRP's HIV Natural History Study Data Set". Retrieved 3 March 2013.
- ↑ Craig, F. E.; Brinkman, R. R.; Eyck, S. T.; Aghaeepour, N. (2013). "Computational analysis optimizes the flow cytometric evaluation for lymphoma". Cytometry Part B: n/a. doi:10.1002/cytob.21115. PMID 23873623.
- ↑ Villanova, F.; Di Meglio, P.; Inokuma, M.; Aghaeepour, N.; Perucha, E.; Mollon, J.; Nomura, L.; Hernandez-Fuentes, M.; Cope, A.; Prevost, A. T.; Heck, S.; Maino, V.; Lord, G.; Brinkman, R. R.; Nestle, F. O. (2013). Von Herrath, Matthias G (ed.). "Integration of Lyoplate Based Flow Cytometry and Computational Analysis for Standardized Immunological Biomarker Discovery". PLOS ONE. 8 (7): e65485. Bibcode:2013PLoSO...865485V. doi:10.1371/journal.pone.0065485. PMC 3701052. PMID 23843942.
- 1 2 Maecker, H. T.; McCoy, J. P.; Nussenblatt, R. (2012). "Standardizing immunophenotyping for the Human Immunology Project". Nature Reviews Immunology. 12 (3): 191–200. doi:10.1038/nri3158. PMC 3409649. PMID 22343568.
- 1 2 3 4 Schadt, E. E.; Linderman, M. D.; Sorenson, J.; Lee, L.; Nolan, G. P. (2010). "Computational solutions to large-scale data management and analysis". Nature Reviews Genetics. 11 (9): 647–657. doi:10.1038/nrg2857. PMC 3124937. PMID 20717155.