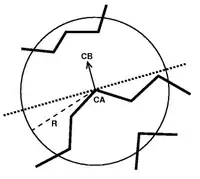
Half Sphere exposure (HSE) is a protein solvent exposure measure that was first introduced by Hamelryck (2005).[1] Like all solvent exposure measures it measures how buried amino acid residues are in a protein. It is found by counting the number of amino acid neighbors within two half spheres of chosen radius around the amino acid. The calculation of HSE is found by dividing a contact number (CN) sphere in two halves by the plane perpendicular to the Cβ-Cα vector. This simple division of the CN sphere results in two strikingly different measures, HSE-up and HSE-down. HSE-up is defined as the number of Cα atoms in the upper half (containing the pseudo-Cβ atom) and analogously HSE-down is defined as the number of Cα atoms in the opposite sphere.
If only Cα atoms are available (as is the case for many simplified representations of protein structure), a related measure, called HSEα, can be used. HSEα uses a pseudo-Cβ instead of the real Cβ atom for its calculation. The position of this pseudo-Cβ atom (pCβ) is derived from the positions of preceding Cα−1 and the following Cα+1. The Cα-pCβ vector is calculated by adding the Cα−1-Cα0 and Cα+1-Cα0 vectors.
HSE is used in predicting discontinuous B-cell epitopes.[2] Song et al. have developed an online webserver termed HSEpred to predict half-sphere exposure from protein primary sequences.[3] HSEpred server can achieve the correlation coefficients of 0.72 and 0.68 between the predicted and observed HSE-up and HSE-down measures, respectively, when evaluated on a well-prepared non-homologous protein structure dataset. Moreover, residue contact number (CN) can also be accurately predicted by HSEpred webserver using the summation of the predicted HSE-up and HSE-down values, which has further enlarged the application of this new solvent exposure measure.
Recently, Heffernan et al. has developed the most accurate predictor for both HSEα and HSEβ based on a big dataset by using multiple-step iterative deep neural-network learning.[4] The predicted HSEa shows a higher correlation coefficient to the stability change by residue mutants than predicted HSEβ and ASA. The results, together with its easy Ca-atom-based calculation, highlight the potential usefulness of predicted HSEa for protein structure prediction and refinement as well as function prediction.
References
- ↑ Hamelryck, T. (2005), "An amino acid has two sides: A new 2D measure provides a different view of solvent exposure", Proteins: Structure, Function, and Bioinformatics, 59 (1): 38–48, CiteSeerX 10.1.1.516.4528, doi:10.1002/prot.20379, PMID 15688434, S2CID 10631851.
- ↑ Sweredoski, Michael J.; Baldi, Pierre (2008), "PEPITO: Improved discontinuous B-cell epitope prediction using multiple distance thresholds and Half Sphere Exposure", Bioinformatics, 24 (12): 1459–1460, doi:10.1093/bioinformatics/btn199, PMID 18443018.
- ↑ Song, J.; Tan, H.; Takemoto, K.; Akutsu, T. (2008), "HSEpred: predict half-sphere exposure from protein sequences", Bioinformatics, 24 (13): 1489–1497, doi:10.1093/bioinformatics/btn222, PMID 18467349.
- ↑ Heffernan, Rhys; et al. (2016), "Highly accurate sequence-based prediction of half-sphere exposures of amino acid residues in proteins", Bioinformatics, 32 (6): 843–9, doi:10.1093/bioinformatics/btv665, PMID 26568622, S2CID 22034498