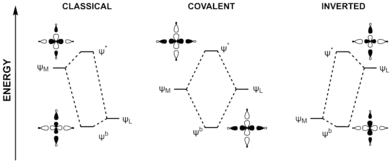
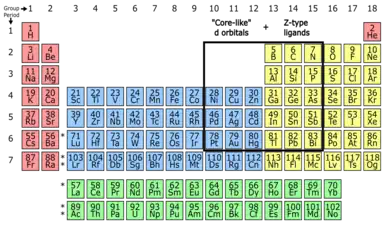
Inverted ligand field theory (ILFT) describes a phenomenon in the bonding of coordination complexes where the lowest unoccupied molecular orbital is primarily of ligand character.[2][1] This is contrary to the traditional ligand field theory or crystal field theory picture and arises from the breaking down of the assumption that in organometallic complexes, ligands are more electronegative and have frontier orbitals below those of the d orbitals of electropositive metals.[3][4] As we move to the right of the d-block and approach the transition-metal - main group boundary, the d orbitals become more core-like, making their cations more electronegative. This decreases their energies and eventually arrives at a point where they are lower in energy than the ligand frontier orbitals.[2] Here the ligand field inverts so that the bonding orbitals are more metal-based, and antibonding orbitals more ligand-based. The relative arrangement of the d orbitals are also inverted in complexes displaying this inverted ligand field.[2] This has consequences in our understanding of accessible metal oxidation states, and the reactivity of complexes exhibiting ILFT.
History
The first example of an inverted ligand field was demonstrated in paper form 1995 by James Snyder.[5] In this theoretical paper, Snyder proposed that the [Cu(CF3)4]- complexes reported by Naumann et al. and assigned a formal oxidation state of 3+ at the copper[6] would be better thought of as Cu(I). By comparing the d-orbital occupation, calculated charges and orbital population of [Cu(CF3)4]- "Cu(III)" complex and the formally Cu(I) [Cu(CH3)2]- complex, they illustrated how the former could be better described as a d10 copper complex experiencing two electron donation from the CF3- ligands.[5] The phenomenon, termed an inverted ligand field by Roald Hoffman, began to be described by Aullón and Alvarez as they identified this phenonmenon as being a result of relative electronegativities.[7] Lancaster and co-workers later provided experimental evidence to support the assignment of this oxidation state. Using UV/visible/near IR spectroscopy, Cu K-edge X-ray absorption spectroscopy, and 1s2p resonant inelastic X-ray scattering in concert with density functional theory, multiplet theory, and multireference calculations, they were able to map the ground state electronic configuration. This showed that the lowest unoccupied orbital was of primarily trifluoromethyl character. This confirmed the presence of an inverted ligand field and started building experimental tools to probe this phenomenon.[8] Since the Snyder case, many other complexes of later transition metals have been shown to display inverted ligand field through both theoretical and experimental methods.
Probing inverted ligand fields
Computational and experimental techniques have been imperative for the study of inverted ligand fields, especially when used in cooperatively.
Computational
Computational methods have played a large role in understanding the nature of bonding in both molecular and solid-state systems displaying inverted ligand fields. The Hoffman group has completed many calculations to probe occurrence of inverted ligand fields in varying systems.[9] In a study of the absorption of CO on PtBi and PtBi2 surfaces, on an octahedral [Pt(BiH3)6]4+ model with a Pt thought of having a formal 4+ oxidation state, the team found that the t2g metal orbitals were higher energy that the eg orbitals. This inversion of the d orbital ordering was attributed to the bismuth based ligands being higher in energy than the metal d orbitals.[10] In another study involving calculations on Ag(III) salt KAgF4, other Ag(II), and Ag(III) compounds, the Ag d orbitals were found to be below those of the fluoride ligand orbitals,[11] and was confirmed by Grochala and cowrokers by core and valence spectroscopies.[12]
The Mealli group developed the program Computer Aided Composition of Atomic Orbitals (CACAO) to provide visualised molecular orbitals analyses based on perturbation theory principles.[13] This program successfully displayed orbital energy inversion with organometallic complexes containing electronegative metals such as Ni or Cu bound to electropositive ligand atoms such as B, Si, or Sn.[13] In these cases the bonding was described as a ligand to metal dative bond or sigma backdonation.[14]

Alvarez and coworkers used computational methods to illustrate ligand field inversion in the band structures of solid state materials. The group found that, contrary to the classical bonding scheme, in calculated MoNiP8 band structures the eg-type orbitals of the octahedral nickel atom were found to be the major component of an occupied band below the t2g set.[16] Additionally, the band around the fermi level which included the Ni+ antibonding orbitals were found to be mostly of phosphorus character, a clear example if an inverted ligand field. Similar observations were made in other solid state materials like the skutterudite CoP3 structure.[17][15] A consequence of the inverted ligand field in this case is that the conductivity in skutterudites is associated with the phosphorus rings rather than the metal atoms.
Experimental
X-ray absorption spectroscopy (XAS) has been a powerful tool in deducing the oxidation states of transition metals.[18] Energy shifts in XAS are higher due to the higher effective nuclear charge of atoms in higher oxidations, presumably due to the higher binding energy for deeper, more core-like electrons.[2]
Despite this being a very powerful technique, competing effects on the rising edge positions can make assignment difficult. It was initially thought that the weak, quadrupole-allowed pre-edge peak assigned as the Cu 1s to 3d transition could be used to distinguish between Cu(II) and Cu(III) with the features appearing at 8979 +/- 0.3 eV and 8981 +/- 0.5 eV, respectively.[19] Ab initio calculations by Tomson, Wieghardt, and co-workers displayed that pre-peaks previously assigned as Cu(III) could be displayed by Cu(II) bearing complexes.[20] Many groups have displayed that metal K-edge XAS transitions involving ligand-localised acceptor orbitals, as well as spectral shifts from change in coordination environment, can make metal K-edge analysis less predictable.[21][22][23][24]
The most sussessful use of K and L-edge XAS provide valuable information on the composition of molecular orbitals and display inverted ligand fields has been done in studies that made use of computational techniques in concert with experimental techniques. This was the case of the L2[Cu2(S2)n]2+ complexes of York, Brown, and Tolman,[25] and the Cu(CF3)4- by various groups including Hoffman,[2] Overgaard,[26] and Lancaster.[1][8]
Another experimental tool used to probe ligand field inversion includes Electron paramagnetic resonance (ESR/EPR), which can provide information regarding the metal electronic configuration, the nature of the SOMO, and high resolution information on the ligands.[27]
Impact of charge and geometry
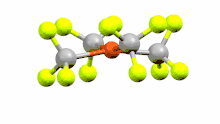
4)n-.png.webp)
Changes in both charge and geometry of organometallic complexes can greatly vary the energies of molecular orbitals and can therefore dictate the likelihood of observing an inverted ligand field. Hoffman and coworkers explored the impact of these variables by calculating the atomic composition of molecular orbitals for mono- di- and trianion copper complexes.[2] The square planar monoanion displayed the reported ligand field inversion. The "Cu(II)" which has an intermediate square planar to tetrahedral geometry also displayed this feature with the antibonding t2-derived orbital being mostly of ligand character and the x2-y2 orbital being the lowest molecular orbital of the d block. The tetrahedral trianion showed a return to the Werner-type ligand field.[2] By modulating the geometry of the "Cu(II)" species and displaying the change in energies of MO on walsh diagrams, the group was able to show how the complex could display both a classical and inverted ligand field when in Td and SP geometry respectively.[2] Additional calculations on the Cu(I) with non-tetrahedral geometry also displayed an inverted ligand field. This indicated the importance of not just oxidation state but geometry in determining the inversion of a ligand field.

Consequences on bonding
The inversion of ligand fields has interesting implications on the nature of reactivity of organometallic complexes. This sigma non-innocence of ligands arising from inverted ligand fields could therefore be used to tune reactivity of complexes and open space in understanding the mechanisms of existing reactions.
In an analysis of the [ZnF4]2- , it was found that due to ligand field inversion displayed in this species, core ionization removes an electron from the metal-rich bonding t2 orbital, lengthening the Zn-F bonds. This is contrary to the classical ligand field where ionization would remove an electron from the antibonding t2 orbital shortening the Zn-F bonds.[2]
The presence of electron-deficient ligands also result in an inverted ligand field. Calculations have shown that the large O 2p contribution into the LUMO/LUMO+1 in [(LTEEDCu)2(O2)]2+ should make the complex highly oxidizing as it contains electron deficient O2- ligands.[1] Studies have corroborated this property as this complex has shown to be able to undergo C-H and C-F activation and aromatic hydroxylation.[28][29][30]
There is evidence showing that reductive elimination on species displaying ligand field inversion do not undergo a redox event at the metal center. The C-CF3 bond formation by "Ni(IV)" complexes[31] was completed without redox participation of the Nickel.[32] The metal appears to remain Ni(II) throughout the reaction. The mechanism is thought to be through the attack of a masked electrophilic cation by anionic CF3. The electron deficiency here is due to the inverted ligand field.[32]
References
- 1 2 3 4 DiMucci, Ida M.; Lukens, James T.; Chatterjee, Sudipta; Carsch, Kurtis M.; Titus, Charles J.; Lee, Sang Jun; Nordlund, Dennis; Betley, Theodore A.; MacMillan, Samantha N.; Lancaster, Kyle M. (2019-11-20). "The Myth of d 8 Copper(III)". Journal of the American Chemical Society. 141 (46): 18508–18520. doi:10.1021/jacs.9b09016. ISSN 0002-7863. PMC 7256958. PMID 31710466.
- 1 2 3 4 5 6 7 8 9 10 11 Hoffmann, Roald; Alvarez, Santiago; Mealli, Carlo; Falceto, Andrés; Cahill, Thomas J.; Zeng, Tao; Manca, Gabriele (2016-07-27). "From Widely Accepted Concepts in Coordination Chemistry to Inverted Ligand Fields". Chemical Reviews. 116 (14): 8173–8192. doi:10.1021/acs.chemrev.6b00251. ISSN 0009-2665. PMID 27398715.
- ↑ Albright, Thomas A. (2013). Orbital interactions in chemistry. Jeremy K. Burdett, Myung-Hwan Whangbo (2nd ed.). Hoboken, New Jersey. ISBN 978-0-471-08039-8. OCLC 823294395.
{{cite book}}: CS1 maint: location missing publisher (link) - ↑ Griffith, J. S.; Orgel, L. E. (1957-01-01). "Ligand-field theory". Quarterly Reviews, Chemical Society. 11 (4): 381–393. doi:10.1039/QR9571100381. ISSN 0009-2681.
- 1 2 Snyder, James (1995). "Distinguishing Copper d8 and d10 Configurations in a Highly Ionic Complex; A Nonformal Metal Oxidation State". Angewandte Chemie International Edition in English. 35 (9): 986–987. doi:10.1002/anie.199509862 – via Wiley.
- ↑ Naumann, Dieter; Roy, Thomas; Tebbe, Karl-Friedrich; Crump, Wolfgang (1993-10-10). "Synthesis and Structure of Surprisingly Stable Tetrakis(trifluoromethyl)cuprate(III) Salts". Angewandte Chemie International Edition in English. 32 (10): 1482–1483. doi:10.1002/anie.199314821. ISSN 0570-0833.
- ↑ Aullón, Gabriel; Alvarez, Santiago (2009-05-01). "Oxidation states, atomic charges and orbital populations in transition metal complexes". Theoretical Chemistry Accounts. 123 (1): 67–73. doi:10.1007/s00214-009-0537-9. ISSN 1432-2234. S2CID 97419872.
- 1 2 Walroth, Richard C.; Lukens, James T.; MacMillan, Samantha N.; Finkelstein, Kenneth D.; Lancaster, Kyle M. (2016-02-17). "Spectroscopic Evidence for a 3d 10 Ground State Electronic Configuration and Ligand Field Inversion in [Cu(CF 3 ) 4 ] 1–". Journal of the American Chemical Society. 138 (6): 1922–1931. doi:10.1021/jacs.5b10819. ISSN 0002-7863. PMID 26844693.
- ↑ Zheng, Chong; Hoffmann, Roald (1989-03-01). "Conjugation in the 3-connected net: the aluminum diboride and thorium disilicide structures and their transition-metal derivatives". Inorganic Chemistry. 28 (6): 1074–1080. doi:10.1021/ic00305a015. ISSN 0020-1669.
- ↑ Oana, Melania; Hoffmann, Roald; Abruña, Héctor D.; DiSalvo, Francis J. (2005-01-01). "Adsorption of CO on PtBi2 and PtBi surfaces". Surface Science. 574 (1): 1–16. Bibcode:2005SurSc.574....1O. doi:10.1016/j.susc.2004.09.045. ISSN 0039-6028.
- ↑ Grochala, Wojciech; Hoffmann, Roald (2010-05-24). "ChemInform Abstract: Real and Hypothetical Intermediate-Valence AgII/AgIII and AgII/AgI Fluoride Systems as Potential Superconductors". ChemInform. 32 (43): no. doi:10.1002/chin.200143247. ISSN 0931-7597.
- ↑ Grochala, Wojciech; Egdell, Russ G.; Edwards, Peter P.; Mazej, Zoran; Žemva, Boris (2003-09-10). "On the Covalency of Silver-Fluorine Bonds in Compounds of Silver(I), Silver(II) and Silver(III)". ChemPhysChem. 4 (9): 997–1001. doi:10.1002/cphc.200300777. ISSN 1439-4235. PMID 14562447.
- 1 2 Mealli, Carlo; Proserpio, Davide M. (1990-05-01). "MO theory made visible". Journal of Chemical Education. 67 (5): 399. Bibcode:1990JChEd..67..399M. doi:10.1021/ed067p399. ISSN 0021-9584.
- ↑ Mealli, Carlo; Ghilardi, Carlo A.; Orlandini, Annabella (1992-11-01). "Structural flexibility and bonding capabilities of the ligand np3 toward the transition metals". Coordination Chemistry Reviews. 120: 361–387. doi:10.1016/0010-8545(92)80059-z. ISSN 0010-8545.
- 1 2 Llunell, Miquel; Alvarez, Santiago; Alemany, Pere (1998). "Skutterudite vs. ReO3 structures for MX3 solids: electronic requirements". Journal of the Chemical Society, Dalton Transactions (7): 1195–1200. doi:10.1039/a708200b. ISSN 0300-9246.
- ↑ LLUNELL, M.; ALVAREZ, S.; ALEMANY, P.; HOFFMANN, R. (2010-08-04). "ChemInform Abstract: Electronic Structure, Bonding, and Electrical Properties of MoNiP8". ChemInform. 27 (46): no. doi:10.1002/chin.199646001. ISSN 0931-7597.
- ↑ Llunell, Miquel; Alemany, Pere; Alvarez, Santiago; Zhukov, Vladlen P.; Vernes, Andreas (1996-04-15). "Electronic structure and bonding in skutterudite-type phosphides". Physical Review B. 53 (16): 10605–10609. Bibcode:1996PhRvB..5310605L. doi:10.1103/physrevb.53.10605. hdl:2445/10836. ISSN 0163-1829. PMID 9982624.
- ↑ Glaser, Thorsten; Hedman, Britt; Hodgson, Keith O.; Solomon, Edward I. (2001-03-06). "ChemInform Abstract: Ligand K-Edge X-Ray Absorption Spectroscopy: A Direct Probe of Ligand-Metal Covalency". ChemInform. 32 (10): no. doi:10.1002/chin.200110300. ISSN 0931-7597.
- ↑ DuBois, Jennifer L.; Mukherjee, Pulakesh; Stack, T. D. P.; Hedman, Britt; Solomon, Edward I.; Hodgson, Keith O. (2000-06-01). "A Systematic K-edge X-ray Absorption Spectroscopic Study of Cu(III) Sites". Journal of the American Chemical Society. 122 (24): 5775–5787. doi:10.1021/ja993134p. ISSN 0002-7863.
- ↑ Tomson, Neil C.; Williams, Kamille D.; Dai, Xuliang; Sproules, Stephen; DeBeer, Serena; Warren, Timothy H.; Wieghardt, Karl (2015). "Re-evaluating the Cu K pre-edge XAS transition in complexes with covalent metal–ligand interactions". Chemical Science. 6 (4): 2474–2487. doi:10.1039/c4sc03294b. ISSN 2041-6520. PMC 5647745. PMID 29308158. S2CID 246748080.
- ↑ "Xray Absorption Spectroscopy as a Probe of Ligand Noninnocence in Metallocorroles: The Case of Copper Corroles". doi:10.1021/acs.inorgchem.9b00128.s001. hdl:10037/18125. Retrieved 2023-03-12.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Walroth, Richard C.; Uebler, Jacob W. H.; Lancaster, Kyle M. (2015). "Probing CuI in homogeneous catalysis using high-energy-resolution fluorescence-detected X-ray absorption spectroscopy". Chemical Communications. 51 (48): 9864–9867. doi:10.1039/c5cc01827g. ISSN 1359-7345. PMC 4540364. PMID 25994112.
- ↑ Carsch, Kurtis M.; DiMucci, Ida M.; Iovan, Diana A.; Li, Alex; Zheng, Shao-Liang; Titus, Charles J.; Lee, Sang Jun; Irwin, Kent D.; Nordlund, Dennis; Lancaster, Kyle M.; Betley, Theodore A. (2019-09-13). "Synthesis of a copper-supported triplet nitrene complex pertinent to copper-catalyzed amination". Science. 365 (6458): 1138–1143. Bibcode:2019Sci...365.1138C. doi:10.1126/science.aax4423. ISSN 0036-8075. PMC 7256962. PMID 31515388.
- ↑ KAU, L.-S.; SPIRA-SOLOMON, D. J.; PENNER-HAHN, J. E.; HODGSON, K. O.; SOLOMON, E. I. (1988-02-09). "ChemInform Abstract: X-Ray Absorption Edge Determination of the Oxidation State and Coordination Number of Copper: Application to the Type 3 Site in Rhus vernicifera Laccase and Its Reaction with Oxygen". ChemInform. 19 (6). doi:10.1002/chin.198806038. ISSN 0931-7597.
- ↑ Sarangi, Ritimukta; York, John T.; Helton, Matthew E.; Fujisawa, Kiyoshi; Karlin, Kenneth D.; Tolman, William B.; Hodgson, Keith O.; Hedman, Britt; Solomon, Edward I. (2008-01-01). "X-Ray Absorption Spectroscopic and Theoretical Studies on (L) 2 [Cu 2 (S 2 ) n ] 2+ Complexes: Disulfide versus Disulfide(•1−) Bonding". Journal of the American Chemical Society. 130 (2): 676–686. doi:10.1021/ja0762745. ISSN 0002-7863. PMC 2570853. PMID 18076173.
- ↑ Overgaard, Jacob (2019-01-15). "Experimental X-ray Electron Density Study of Atomic Charges, Oxidation States, and Inverted Ligand Field in Cu(CF3)4–". Inorganic Chemistry. 58 (3): 2133–2139. doi:10.1021/acs.inorgchem.8b03226. hdl:2434/971202. PMID 30645110. S2CID 58640050.
- ↑ Kisgeropoulos, Effie C.; Manesis, Anastasia C.; Shafaat, Hannah S. (2021-01-20). "Ligand Field Inversion as a Mechanism to Gate Bioorganometallic Reactivity: Investigating a Biochemical Model of Acetyl CoA Synthase Using Spectroscopy and Computation". Journal of the American Chemical Society. 143 (2): 849–867. doi:10.1021/jacs.0c10135. ISSN 0002-7863. PMID 33415980. S2CID 231195483.
- ↑ Mirica, Liviu M.; Ottenwaelder, Xavier; Stack, T. Daniel P. (2004-05-11). "Structure and Spectroscopy of Copper—Dioxygen Complexes". ChemInform. 35 (19). doi:10.1002/chin.200419297. ISSN 0931-7597.
- ↑ Company, Anna; Palavicini, Sara; Garcia-Bosch, Isaac; Mas-Ballesté, Ruben; Que, Lawrence; Rybak-Akimova, Elena V.; Casella, Luigi; Ribas, Xavi; Costas, Miquel (2008-04-18). "Tyrosinase-Like Reactivity in a CuIII2(μ-O)2 Species". Chemistry - A European Journal. 14 (12): 3535–3538. doi:10.1002/chem.200800229. ISSN 0947-6539. PMID 18348133.
- ↑ Serrano-Plana, Joan; Garcia-Bosch, Isaac; Miyake, Ryosuke; Costas, Miquel; Company, Anna (2014-09-01). "Selective Ortho -Hydroxylation-Defluorination of 2-Fluorophenolates with a Bis(μ-oxo)dicopper(III) Species". Angewandte Chemie International Edition. 53 (36): 9608–9612. doi:10.1002/anie.201405060. PMID 25044926.
- ↑ Bour, James R.; Camasso, Nicole M.; Sanford, Melanie S. (2015-06-16). "Oxidation of Ni(II) to Ni(IV) with Aryl Electrophiles Enables Ni-Mediated Aryl–CF3 Coupling". Journal of the American Chemical Society. 137 (25): 8034–8037. doi:10.1021/jacs.5b04892. ISSN 0002-7863. PMID 26079544.
- 1 2 Steen, Jelte S.; Knizia, Gerald; Klein, Johannes E. M. N. (2019-07-15). "σ‐Noninnocence: Masked Phenyl‐Cation Transfer at Formal Ni(IV)". Angewandte Chemie. 131 (37): 13267–13273. doi:10.1002/ange.201906658. ISSN 0044-8249. S2CID 241689909.