| Oxidation state | Representative compound |
|---|---|
| −4 (d10s2) | [FeIn6−xSnx][1] |
| −2 (d10) | Disodium tetracarbonylferrate (Collman's reagent) |
| −1 (d9) | Fe 2(CO)2− 8 |
| 0 (d8) | Iron pentacarbonyl |
| 1 (d7) | Cyclopentadienyliron dicarbonyl dimer ("Fp2") |
| 2 (d6) | Ferrous sulfate, ferrocene |
| 3 (d5) | Ferric chloride, ferrocenium tetrafluoroborate |
| 4 (d4) | Fe(diars) 2Cl2+ 2, Ferryl tetrafluoroborate |
| 5 (d3) | FeO3− 4 |
| 6 (d2) | Potassium ferrate |
| 7 (d1) | [FeO4]– (matrix isolation, 4K) |
Iron shows the characteristic chemical properties of the transition metals, namely the ability to form variable oxidation states differing by steps of one and a very large coordination and organometallic chemistry: indeed, it was the discovery of an iron compound, ferrocene, that revolutionalized the latter field in the 1950s.[2] Iron is sometimes considered as a prototype for the entire block of transition metals, due to its abundance and the immense role it has played in the technological progress of humanity.[3] Its 26 electrons are arranged in the configuration [Ar]3d64s2, of which the 3d and 4s electrons are relatively close in energy, and thus it can lose a variable number of electrons and there is no clear point where further ionization becomes unprofitable.[4]
Iron forms compounds mainly in the oxidation states +2 (iron(II), "ferrous") and +3 (iron(III), "ferric"). Iron also occurs in higher oxidation states, e.g. the purple potassium ferrate (K2FeO4), which contains iron in its +6 oxidation state. Although iron(VIII) oxide (FeO4) has been claimed, the report could not be reproduced and such a species from the removal of all electrons of the element beyond the preceding inert gas configuration (at least with iron in its +8 oxidation state) has been found to be improbable computationally.[5] However, one form of anionic [FeO4]– with iron in its +7 oxidation state, along with an iron(V)-peroxo isomer, has been detected by infrared spectroscopy at 4 K after cocondensation of laser-ablated Fe atoms with a mixture of O2/Ar.[6] Iron(IV) is a common intermediate in many biochemical oxidation reactions.[7][8] Numerous organoiron compounds contain formal oxidation states of +1, 0, −1, or even −2. The oxidation states and other bonding properties are often assessed using the technique of Mössbauer spectroscopy.[9] Many mixed valence compounds contain both iron(II) and iron(III) centers, such as magnetite and Prussian blue (Fe4(Fe[CN]6)3).[8] The latter is used as the traditional "blue" in blueprints.[10]
Iron is the first of the transition metals that cannot reach its group oxidation state of +8, although its heavier congeners ruthenium and osmium can, with ruthenium having more difficulty than osmium.[11] Ruthenium exhibits an aqueous cationic chemistry in its low oxidation states similar to that of iron, but osmium does not, favoring high oxidation states in which it forms anionic complexes.[11] In the second half of the 3d transition series, vertical similarities down the groups compete with the horizontal similarities of iron with its neighbors cobalt and nickel in the periodic table, which are also ferromagnetic at room temperature and share similar chemistry. As such, iron, cobalt, and nickel are sometimes grouped together as the iron triad.[3]
Unlike many other metals, iron does not form amalgams with mercury. As a result, mercury is traded in standardized 76 pound flasks (34 kg) made of iron.[12]
Iron is by far the most reactive element in its group; it is pyrophoric when finely divided and dissolves easily in dilute acids, giving Fe2+. However, it does not react with concentrated nitric acid and other oxidizing acids due to the formation of an impervious oxide layer, which can nevertheless react with hydrochloric acid.[11] High purity iron, called electrolytic iron, is considered to be resistant to rust, due to its oxide layer.
Binary compounds
Oxides and hydroxides
_oxide.jpg.webp)
-oxide-sample.jpg.webp)

Iron forms various oxide and hydroxide compounds; the most common are iron(II,III) oxide (Fe3O4), and iron(III) oxide (Fe2O3). Iron(II) oxide also exists, though it is unstable at room temperature. Despite their names, they are actually all non-stoichiometric compounds whose compositions may vary.[13] These oxides are the principal ores for the production of iron (see bloomery and blast furnace). They are also used in the production of ferrites, useful magnetic storage media in computers, and pigments. The best known sulfide is iron pyrite (FeS2), also known as fool's gold owing to its golden luster.[8] It is not an iron(IV) compound, but is actually an iron(II) polysulfide containing Fe2+ and S2−
2 ions in a distorted sodium chloride structure.[13]
Halides
_chloride_hexahydrate.jpg.webp)
The binary ferrous and ferric halides are well-known. The ferrous halides typically arise from treating iron metal with the corresponding hydrohalic acid to give the corresponding hydrated salts.[8]
- Fe + 2 HX → FeX2 + H2 (X = F, Cl, Br, I)
Iron reacts with fluorine, chlorine, and bromine to give the corresponding ferric halides, ferric chloride being the most common.[14]
- 2 Fe + 3 X2 → 2 FeX3 (X = F, Cl, Br)
Ferric iodide is an exception, being thermodynamically unstable due to the oxidizing power of Fe3+ and the high reducing power of I−:[14]
- 2 I− + 2 Fe3+ → I2 + 2 Fe2+ (E0 = +0.23 V)
Ferric iodide, a black solid, is not stable in ordinary conditions, but can be prepared through the reaction of iron pentacarbonyl with iodine and carbon monoxide in the presence of hexane and light at the temperature of −20 °C, with oxygen and water excluded.[14] Complexes of ferric iodide with some soft bases are known to be stable compounds.[15][16]
Solution chemistry

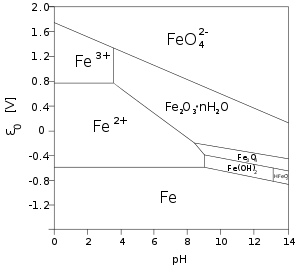
The standard reduction potentials in acidic aqueous solution for some common iron ions are given below:[11]
| Fe2+ + 2 e− | ⇌ Fe | E0 = −0.447 V |
| Fe3+ + 3 e− | ⇌ Fe | E0 = −0.037 V |
| FeO2− 4 + 8 H+ + 3 e− | ⇌ Fe3+ + 4 H2O | E0 = +2.20 V |
The red-purple tetrahedral ferrate(VI) anion is such a strong oxidizing agent that it oxidizes nitrogen and ammonia at room temperature, and even water itself in acidic or neutral solutions:[14]
- 4 FeO2−
4 + 10 H
2O → 4 Fe3+
+ 20 OH−
+ 3 O2
The Fe3+ ion has a large simple cationic chemistry, although the pale-violet hexaquo ion [Fe(H2O)6]3+ is very readily hydrolyzed when pH increases above 0 as follows:[17]
| [Fe(H2O)6]3+ | ⇌ [Fe(H2O)5(OH)]2+ + H+ | K = 10−3.05 mol dm−3 |
| [Fe(H2O)5(OH)]2+ | ⇌ [Fe(H2O)4(OH)2]+ + H+ | K = 10−3.26 mol dm−3 |
| 2[Fe(H2O)6]3+ | ⇌ [Fe(H2O)4(OH)]4+2 + 2H+ + 2H2O | K = 10−2.91 mol dm−3 |
-sulfate-heptahydrate-sample.jpg.webp)
As pH rises above 0 the above yellow hydrolyzed species form and as it rises above 2–3, reddish-brown hydrous iron(III) oxide precipitates out of solution. Although Fe3+ has a d5 configuration, its absorption spectrum is not like that of Mn2+ with its weak, spin-forbidden d–d bands, because Fe3+ has higher positive charge and is more polarizing, lowering the energy of its ligand-to-metal charge transfer absorptions. Thus, all the above complexes are rather strongly colored, with the single exception of the hexaquo ion – and even that has a spectrum dominated by charge transfer in the near ultraviolet region.[17] On the other hand, the pale green iron(II) hexaquo ion [Fe(H2O)6]2+ does not undergo appreciable hydrolysis. Carbon dioxide is not evolved when carbonate anions are added, which instead results in white iron(II) carbonate being precipitated out. In excess carbon dioxide this forms the slightly soluble bicarbonate, which occurs commonly in groundwater, but it oxidises quickly in air to form iron(III) oxide that accounts for the brown deposits present in a sizeable number of streams.[18]
Coordination compounds
Due to its electronic structure, iron has a very large coordination and organometallic chemistry.

Many coordination compounds of iron are known, although few exhibit high-valent iron. A typical six-coordinate anion is hexachloroferrate(III), [FeCl6]3−, found in the mixed salt tetrakis(methylammonium) hexachloroferrate(III) chloride.[19][20] Complexes with multiple bidentate ligands have geometric isomers. For example, the trans-chlorohydridobis(bis-1,2-(diphenylphosphino)ethane)iron(II) complex is used as a starting material for compounds with the Fe(dppe)2 moiety.[21][22] The ferrioxalate ion with three oxalate ligands (shown at right) displays helical chirality with its two non-superposable geometries labelled Λ (lambda) for the left-handed screw axis and Δ (delta) for the right-handed screw axis, in line with IUPAC conventions.[17] Potassium ferrioxalate is used in chemical actinometry and along with its sodium salt undergoes photoreduction applied in old-style photographic processes. The dihydrate of iron(II) oxalate has a polymeric structure with co-planar oxalate ions bridging between iron centres with the water of crystallisation located forming the caps of each octahedron, as illustrated below.[23]
(H2O)2-chain-from-xtal-2008-CM-3D-balls.png.webp)
iron(III)_chloride.jpg.webp)
Iron(III) complexes are quite similar to those of chromium(III) with the exception of iron(III)'s preference for O-donor instead of N-donor ligands. The latter tend to be rather more unstable than iron(II) complexes and often dissociate in water. Many Fe–O complexes show intense colors and are used as tests for phenols or enols. For example, in the ferric chloride test, used to determine the presence of phenols, iron(III) chloride reacts with a phenol to form a deep violet complex:[17]
- 3 ArOH + FeCl3 → Fe(OAr)3 + 3 HCl (Ar = aryl)
Among the halide and pseudohalide complexes, fluoro complexes of iron(III) are the most stable, with the colorless [FeF5(H2O)]2− being the most stable in aqueous solution. Chloro complexes are less stable and favor tetrahedral coordination as in [FeCl4]−; [FeBr4]− and [FeI4]− are reduced easily to iron(II). Thiocyanate is a common test for the presence of iron(III) as it forms the blood-red [Fe(SCN)(H2O)5]2+. Like manganese(II), most iron(III) complexes are high-spin, the exceptions being those with ligands that are high in the spectrochemical series such as cyanide. An example of a low-spin iron(III) complex is [Fe(CN)6]3−. The cyanide ligands may easily be detached in [Fe(CN)6]3−, and hence this complex is poisonous, unlike the iron(II) complex [Fe(CN)6]4− found in Prussian blue,[17] which does not release hydrogen cyanide except when dilute acids are added.[18] Iron shows a great variety of electronic spin states, including every possible spin quantum number value for a d-block element from 0 (diamagnetic) to 5⁄2 (5 unpaired electrons). This value is always half the number of unpaired electrons. Complexes with zero to two unpaired electrons are considered low-spin and those with four or five are considered high-spin.[13]
Iron(II) complexes are less stable than iron(III) complexes but the preference for O-donor ligands is less marked, so that for example [Fe(NH3)6]2+ is known while [Fe(NH3)6]3+ is not. They have a tendency to be oxidized to iron(III) but this can be moderated by low pH and the specific ligands used.[18]
Organometallic compounds

carbonyl
Organoiron chemistry is the study of organometallic compounds of iron, where carbon atoms are covalently bound to the metal atom. They are many and varied, including cyanide complexes, carbonyl complexes, sandwich and half-sandwich compounds.
Prussian blue or "ferric ferrocyanide", Fe4[Fe(CN)6]3, is an old and well-known iron-cyanide complex, extensively used as pigment and in several other applications. Its formation can be used as a simple wet chemistry test to distinguish between aqueous solutions of Fe2+ and Fe3+ as they react (respectively) with potassium ferricyanide and potassium ferrocyanide to form Prussian blue.[8]
Another old example of an organoiron compound is iron pentacarbonyl, Fe(CO)5, in which a neutral iron atom is bound to the carbon atoms of five carbon monoxide molecules. The compound can be used to make carbonyl iron powder, a highly reactive form of metallic iron. Thermolysis of iron pentacarbonyl gives triiron dodecacarbonyl, Fe3(CO)12, a complex with a cluster of three iron atoms at its core. Collman's reagent, disodium tetracarbonylferrate, is a useful reagent for organic chemistry; it contains iron in the −2 oxidation state. Cyclopentadienyliron dicarbonyl dimer contains iron in the rare +1 oxidation state.[24]

.JPG.webp)
A landmark in this field was the discovery in 1951 of the remarkably stable sandwich compound ferrocene Fe(C5H5)2, by Pauson and Kealy[25] and independently by Miller and colleagues,[26] whose surprising molecular structure was determined only a year later by Woodward and Wilkinson[27] and Fischer.[28] Ferrocene is still one of the most important tools and models in this class.[29]
Iron-centered organometallic species are used as catalysts. The Knölker complex, for example, is a transfer hydrogenation catalyst for ketones.[30]
Industrial uses
The iron compounds produced on the largest scale in industry are iron(II) sulfate (FeSO4·7H2O) and iron(III) chloride (FeCl3). The former is one of the most readily available sources of iron(II), but is less stable to aerial oxidation than Mohr's salt ((NH4)2Fe(SO4)2·6H2O). Iron(II) compounds tend to be oxidized to iron(III) compounds in the air.[8]
See also
References
- ↑ Fe(−4), Ru(−4), and Os(−4) have been observed in metal-rich compounds containing octahedral complexes [MIn6−xSnx]; Pt(−3) (as a dimeric anion [Pt–Pt]6−), Cu(−2), Zn(−2), Ag(−2), Cd(−2), Au(−2), and Hg(−2) have been observed (as dimeric and monomeric anions; dimeric ions were initially reported to be [T–T]2− for Zn, Cd, Hg, but later shown to be [T–T]4− for all these elements) in La2Pt2In, La2Cu2In, Ca5Au3, Ca5Ag3, Ca5Hg3, Sr5Cd3, Ca5Zn3(structure (AE2+)5(T–T)4−T2−⋅4e−), Yb3Ag2, Ca5Au4, and Ca3Hg2; Au(–3) has been observed in ScAuSn and in other 18-electron half-Heusler compounds. See Changhoon Lee; Myung-Hwan Whangbo (2008). "Late transition metal anions acting as p-metal elements". Solid State Sciences. 10 (4): 444–449. Bibcode:2008SSSci..10..444K. doi:10.1016/j.solidstatesciences.2007.12.001. and Changhoon Lee; Myung-Hwan Whangbo; Jürgen Köhler (2010). "Analysis of Electronic Structures and Chemical Bonding of Metal-rich Compounds. 2. Presence of Dimer (T–T)4– and Isolated T2– Anions in the Polar Intermetallic Cr5B3-Type Compounds AE5T3 (AE = Ca, Sr; T = Au, Ag, Hg, Cd, Zn)". Zeitschrift für Anorganische und Allgemeine Chemie. 636 (1): 36–40. doi:10.1002/zaac.200900421.
- ↑ Greenwood and Earnshaw, p. 905
- 1 2 Greenwood and Earnshaw, p. 1070
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8. (p. 1074).
- ↑ Huang, Wei; Xu, Wen-Hua; Schwarz, W.H.E.; Li, Jun (2016-05-02). "On the Highest Oxidation States of Metal Elements in MO4 Molecules (M = Fe, Ru, Os, Hs, Sm, and Pu)". Inorganic Chemistry. 55 (9): 4616–25. doi:10.1021/acs.inorgchem.6b00442. PMID 27074099.
- ↑ Lu, Jun-Bo; Jian, Jiwen; Huang, Wei; Lin, Hailu; Li, Jun; Zhou, Mingfei (2016-11-16). "Experimental and theoretical identification of the Fe(VII) oxidation state in FeO4−". Phys. Chem. Chem. Phys. 18 (45): 31125–31131. Bibcode:2016PCCP...1831125L. doi:10.1039/c6cp06753k. PMID 27812577.
- ↑ Nam, Wonwoo (2007). "High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions" (PDF). Accounts of Chemical Research. 40 (7): 522–531. doi:10.1021/ar700027f. PMID 17469792. Archived from the original (PDF) on 15 June 2021. Retrieved 22 February 2022.
- 1 2 3 4 5 6 Holleman, Arnold F.; Wiberg, Egon; Wiberg, Nils (1985). "Iron". Lehrbuch der Anorganischen Chemie (in German) (91–100 ed.). Walter de Gruyter. pp. 1125–46. ISBN 3-11-007511-3.
- ↑ Reiff, William Michael; Long, Gary J. (1984). "Mössbauer Spectroscopy and the Coordination Chemistry of Iron". Mössbauer spectroscopy applied to inorganic chemistry. Springer. pp. 245–83. ISBN 978-0-306-41647-7.
- ↑ Ware, Mike (1999). "An introduction in monochrome". Cyanotype: the history, science and art of photographic printing in Prussian blue. NMSI Trading Ltd. pp. 11–19. ISBN 978-1-900747-07-3.
- 1 2 3 4 Greenwood and Earnshaw, pp. 1075–79
- ↑ Gmelin, Leopold (1852). "Mercury and Iron". Hand-book of chemistry. Vol. 6. Cavendish Society. pp. 128–29.
- 1 2 3 Greenwood and Earnshaw, p. 1079
- 1 2 3 4 Greenwood and Earnshaw, pp. 1082–84
- ↑ Siegfried Pohl, Ulrich Bierbach, Wolfgang Saak; "FeI3SC(NMe2)2, a Neutral Thiourea Complex of Iron(III) Iodide", Angewandte Chemie International Edition in English (1989) 28 (6), 776-777. https://doi.org/10.1002/anie.198907761
- ↑ Nicholas A. Barnes, Stephen M.Godfrey, Nicholas Ho, Charles A.McAuliffe, Robin G.Pritchard; "Facile synthesis of a rare example of an iron(III) iodide complex, [FeI3(AsMe3)2], from the reaction of Me3AsI2 with unactivated iron powder", Polyhedron (2013) 55, 67-72. https://doi.org/10.1016/j.poly.2013.02.066
- 1 2 3 4 5 Greenwood and Earnshaw, pp. 1088–91
- 1 2 3 Greenwood and Earnshaw, pp. 1091–97
- ↑ Clausen, C.A.; Good, M.L. (1968). "Stabilization of the hexachloroferrate(III) anion by the methylammonium cation". Inorganic Chemistry. 7 (12): 2662–63. doi:10.1021/ic50070a047.
- ↑ James, B.D.; Bakalova, M.; Lieseganga, J.; Reiff, W.M.; Hockless, D.C.R.; Skelton, B.W.; White, A.H. (1996). "The hexachloroferrate(III) anion stabilized in hydrogen bonded packing arrangements. A comparison of the X-ray crystal structures and low temperature magnetism of tetrakis(methylammonium) hexachloroferrate(III) chloride (I) and tetrakis(hexamethylenediammonium) hexachloroferrate(III) tetrachloroferrate(III) tetrachloride (II)". Inorganica Chimica Acta. 247 (2): 169–74. doi:10.1016/0020-1693(95)04955-X.
- ↑ Giannoccaro, P.; Sacco, A. (1977). "Bis[Ethylenebis(Diphenylphosphine)]‐Hydridoiron Complexes". Inorg. Synth. 17: 69–72. doi:10.1002/9780470132487.ch19. ISBN 978-0-470-13248-7.
- ↑ Lee, J.; Jung, G.; Lee, S.W. (1998). "Structure of trans-chlorohydridobis(diphenylphosphinoethane)iron(II)". Bull. Korean Chem. Soc. 19 (2): 267–69. doi:10.1007/BF02698412. S2CID 35665289.
- ↑ Echigo, Takuya; Kimata, Mitsuyoshi (2008). "Single-crystal X-ray diffraction and spectroscopic studies on humboldtine and lindbergite: weak Jahn–Teller effect of Fe2+ ion". Phys. Chem. Minerals. 35 (8): 467–75. Bibcode:2008PCM....35..467E. doi:10.1007/s00269-008-0241-7. S2CID 98739882.
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1984). Chemistry of the Elements. Oxford: Pergamon Press. pp. 1282–86. ISBN 978-0-08-022057-4..
- ↑ Kealy, T.J.; Pauson, P.L. (1951). "A New Type of Organo-Iron Compound". Nature. 168 (4285): 1039–40. Bibcode:1951Natur.168.1039K. doi:10.1038/1681039b0. S2CID 4181383.
- ↑ Miller, S. A.; Tebboth, J. A.; Tremaine, J. F. (1952). "114. Dicyclopentadienyliron". J. Chem. Soc.: 632–635. doi:10.1039/JR9520000632.
- ↑ Wilkinson, G.; Rosenblum, M.; Whiting, M. C.; Woodward, R. B. (1952). "The Structure of Iron Bis-Cyclopentadienyl". J. Am. Chem. Soc. 74 (8): 2125–2126. doi:10.1021/ja01128a527.
- ↑ Okuda, Jun (2016-12-28). "Ferrocene – 65 Years After". European Journal of Inorganic Chemistry. 2017 (2): 217–219. doi:10.1002/ejic.201601323. ISSN 1434-1948.
- ↑ Greenwood and Earnshaw, p. 1104
- ↑ Bullock, R.M. (11 September 2007). "An Iron Catalyst for Ketone Hydrogenations under Mild Conditions". Angew. Chem. Int. Ed. 46 (39): 7360–63. doi:10.1002/anie.200703053. PMID 17847139.