The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized[1] to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton (which is an enol). It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.;[2] practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.
Theory
It is well-known that Lewis acids and bases can influence carbonyl activity by either protonation of the oxygen or de-protonation of a beta-site to influence electrophilicity at the carbon. In the former case, there is a stabilization effect and priming for a potential leaving group by providing an equilibrium between a formal positive charge and an alcohol group. In either case, the carbon sees enhanced reactivity as the bonds are strained to equalize charge by seeking a nucleophile to which the building charge will equalize into in the subsequent reaction. Potentiation in such reactions is therefore generally driven by how well a material can produce and stabilize a charge gradient through resonance or inductive effects in substituents. The surfaces of acids can catalyze these effects by stabilizing the distortion of the carbonyl's electron cloud, leading to sensitivity in the reaction to acid-base conditions, with acidic conditions typically favoring reaction by enhancing carbonyl leavability.
Mannich reactions are named after their pioneer, Carl Mannich. After the discovery of Mannich reaction in 1912, he found that combination of a ketone, aldehyde and amine consistently produced an addition of the aldehyde unto the site originally containing an acidic β-proton in the ketone. He theorized the mechanism to be one of mixed-aldol; featuring the dehydration of an alcohol and Michael-Addition of the complex. The reaction suffered from a lack of specificity, and this problem persisted until 1997, in which an asymmetrical method was discovered independently by several researchers. Kobayashi[3] et al. used Brønsted-acid to demonstrate that the organic reaction could occur in an aqueous medium, and also found an enantiomeric excess of nearly 55%. It was hypothesized that one of the reactants must be using the acid surface to change its electronic structure since otherwise the mixture would be immiscible, though the study used chiral Lewis acid as catalyst. Prior, Hajos[4] et al. demonstrated a method by which L-proline could be used in an aldol-cyclization.
Subsequent studies have focused on improving the catalyst or materials through substituent effects, from which controls using sterics or restriction of charge sites are expected to improve catalytic yields. Alternatives to the catalyst are not readily explored due to the ubiquity of L-proline, its low cost, and its high selectivity; cementing its proliferation as a catalytic standard. L-proline restricted reactions through its five-membered ring which favours the addition of another reactant in a fixed direction. List[2] et al. theorize in their study that this restriction is primarily in the carboxylic-acid group next to the enamine attachment, which can stabilize the imine product. List also expands upon the role of the imine by listing its ratio with the aldehyde to be 1 using 1H-NMR methods, indicating that the Michael Addition is rate-determining and not the proline-complexation or, obviously, aldehyde activation. This is further confirmed in a Hammett study on para-substituted aromatic aldehydes, in which List confirmed positive correlation between withdrawing effect and increasing imine reactivity, a sign that Michael addition had to be happening with the former aldehyde as an electrophile.[2]
Substituent studies
Substituent studies have focused on increasing or decreasing steric or electronic influences on material to favor product. In 2012,[5] the first non-aromatic ketiminoester was produced with the capability of reduction into either syn- or anti- lactones with NaBH4 reduction. Kano et al. demonstrated a 20:1 excess of desired product, electrophilic enhancement of the ketone by different flanking esters.
Endocyclic ketimines without electron-withdrawing substituents
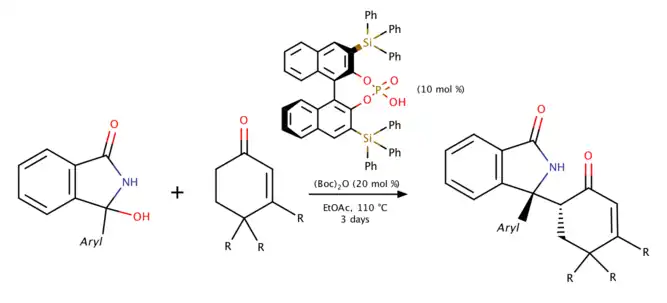
BINOL phosphoric acid-catalyzed Mannich reaction (2019)
Reddy and coworkers proposed the method to produce endocyclic N-acyl ketimines from a stable precursor, 3-aryl-3-hydroxyisoindolin-1-ones in 2019. The reaction yields a high stereoselectivity under high temperature as the adjacent quaternary and stereogenic centers creating chirality.[6] Meanwhile, if 3-hydroxy-3-pentylisoindoline-1-one is used, 95% enamide would be generated instead of Mannich product under the same mechanism.
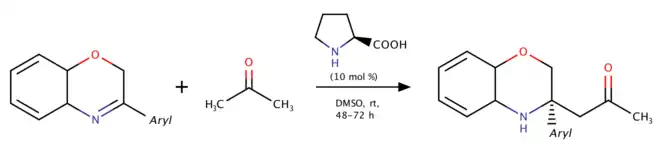
Proline-catalyzed direct asymmetric Mannich reaction of 3-substituted-2H-1,4-benzoxazines (2013)
In 2013, Wang and coworkers studied using 3,4-dihydro-2H-1,4-benzoxazines to produce N-heterocyclic products.[7] This reaction was the first catalytic asymmetric Mannich reaction of 3,4-dihydro-2H-1,4-benzoxazines. The aromatic ring strain in 3,4-dihydro-2H-1,4-benzoxazines helps increase reactivity of the C=N double bond and gives the considerable yield with no electron withdrawing substituents. Similar mechanism also occurred in the reaction catalyzed by wheat germ lipase in 2016 provided by Guan, He and coworkers.[8]
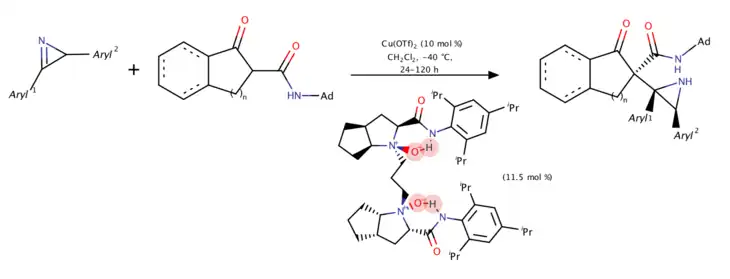
Copper-catalyzed asymmetric Mannich reaction of 2H-azirines with β-keto amides (2018)
In 2018, Lin, Feng, and coworkers developed the first copper-catalyzed catalytic enantioselective Mannich reaction using 2H-azirines and β-ketoamides.[9] The racemic 2H-azirines was first applied and one enantiomer of the 2H-azirine would react with chiral Cu-enolated complex to help overcome its three neighbouring stereogenic centers and gave proper products.
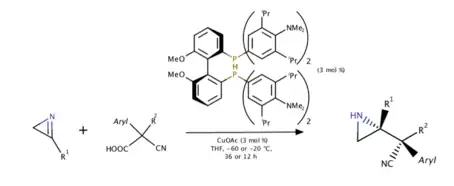
Copper(I)-catalyzed asymmetric decarboxylative Mannich reaction of 2H-azirines (2019)
In 2019, Yin and coworkers used 2H-azirines as an electrophile and a base at the same time to deprotonate cyanoacetic acid and thus a further decarboxylation could proceed. Proton transfer strategy was also discussed since the protonated 2H-azirine obtains high electrophilicity while nonprotonated ones does not even react with exact same environment. Various substituents on both nucleophile and electrophile gave high enantiomer-enriched yields of azirines.[10]
Zn-ProPhenol catalyzed asymmetric Mannich reaction of 2H-azirines with alkynyl cycloalkyl ketones (2020)
In 2020, Trost and coworker proposed a Mannich reaction of 2H-aziridines with alkylnyl cycloalkyl ketones under a bimetallic Zn-ProPhenol complex as the catalyst. The bimetallic Zn-ProPhenol complex employed in the reaction to activate both nucleophile and electrophile since the compound obtains a Bronsted basic site and a Lewis acidic group at chiral center at the same time. The hydrogen bonding between N from aziridine and H from carbonyl group might also contribute chirality centred at nitrogen atom. In this case, N-H bond acetylation product was also generated from the reaction.[11]
Chiral phosphoric acid-catalyzed Mukaiyama–Mannich reaction of endocyclic N-acyl ketimines (2020)
In 2020, Zhang, Ma, and coworkers used 3-hydroxyisoindolin-1-ones to produce endocyclic N-acyl ketimines by chiral phosphoric acid-catalyzed Mukaiyama–Mannich reaction. Difluorinated silyl enol ethers are involved to produce enantioenriched fluoroalkyl-functionalized isoindolones first. Through the reaction under different conditions, two features were discovered. Firstly, hexafluoroisopropyl alcohol could enhance the reactivity and enantioselectivity. Secondly, reaction is better catalyzed by catalyst with trifluoromethyl group chiral barriers.[12]
Isatin-derived ketimines
Isatin-derived ketimine commonly undergoes catalytic asymmetric Mannich reaction to generate 3-substituted-3-aminooxindoles. The structure could be found in various natural or biological molecules, and such reaction could lead to enantio-enriched products. Moreover, the isatin-derived ketimines are comparatively reactive as an electrophile and thus was commonly used in various methods or reactions.
Morimoto, Ohshima, and coworkers applied decarboxylative Mannich reaction on N-unprotected isatin-derived ketimines in 2018.[13] The researchers applied an enantio-enriched chiral oxindoles on the 3-positions with primary amines. In this case, substrates with less substituted groups are more likely to undergo Mannich reactions with higher yield. Also, this routine allows a great range of ketoacids to react with ketimines before decarboxylation and generate Mannich products.

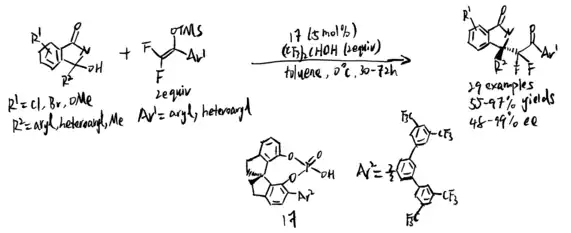
Wolf and coworkers further developed a copper-catalyzed stereo-divergent Mannich reaction in 2020.[14] Isatin-derived ketimines first react with α-fluoro-α-arylnitrile and result in a chiral cuprous keteniminate complex. Then, BTMG would act as a nucleophile and the stereoselectivity would be determined by the chosen of chiral ligands. From experiment, Segphos–copper complex generated anti diastereomers while Taniaphos complex results in syn diastereomers.
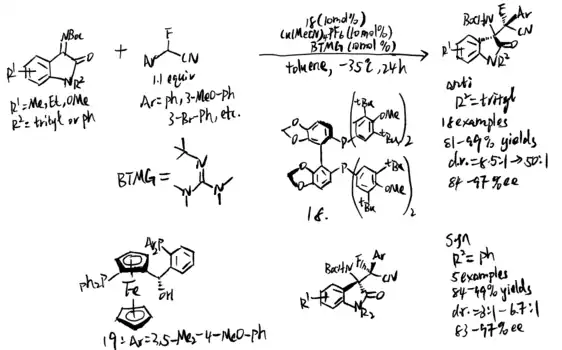
Feng and coworkers used chiral N,N' -dioxide/Zn(II) complexes to catalyze silyl enol ethers and ketimines in 2019.[15] The reaction started with an α-alkenyl addition of silyl enol ethers. Dioxide (compound 21 and 22 in scheme 20) was proved to the best reaction condition for a wide range of isatin-derived ketimines to generate great yield of products with high stereoselectivity. Pyrazolinone-derived ketimines are also found to be suitable substates. The mechanism demonstrates that Mukaiyama–Mannich addition intermediates produced β-amino silyl enol ethers as product.
Acyclic ketimines with electron-withdrawing groups and/or alkynes
Esters, perfluorinated alkyl, or alkyne groups are good electrophiles, that can be used to form ketimine enolate with different nucleophiles. However, subtle differences in ketimine structures can significantly affect stereoselectivities and yields. Nevertheless, many medicinally and synthetically significant chiral structure can be created by creative design on modified ketimines.
In 2016, a novel direct asymmetric Mannich reaction of ketiminoesters with thionolactones were proposed by Terada et al. by using bis(guanidino)iminophosphorane as a chiral organosuperbase catalyst. (scheme 21)[16] Comparing to thionolactones as an appropriate nucleophile, lactone is not acidic enough to be enolized by base catalyst, which makes lactone unable to act as a nucleophile. For electrophiles, having proper substituents on benzoyl protecting group can improve yields and diastereoselectivity. For instance, comparing to trifluromethyl group on ketiminoester, methoxy substituents gives much lower yield and selectivity.

In 2016, direct asymmetric catalytic Mannich reaction of an alpha, beta-unsaturated gamma-butyrolactam with ketiminoesters was proposed by Kumagai el al. using soft Lewis acid/hard Brønsted base cooperative catalysis. (scheme 22)[17] As a result, alpha-addition products were synthesized from alpha,beta-unsaturated gamma-butyrolactams onto ketimines through asymmetric catalysis, which is a novel catalytic asymmetric reaction. The corresponding phosphinoyl protected ketiminoester did not react without the cooperative catalysis by the soft Lewis acid / hard Bronsted base, which means the interaction between copper catalyst (as soft Lewis acid) and thiophosphinoyl protecting group of ketimines (as soft Lewis base) is significant for this successful Mannich reaction.
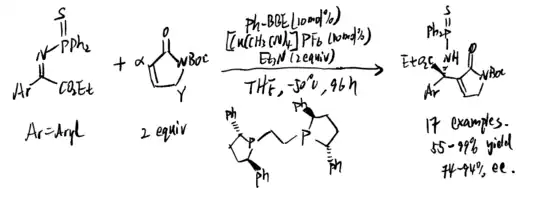
In 2017, a direct enantioselective Mannich reaction with an N-unprotected trifluoromethyl ketiminoester was proposed by Morimoto et al. (scheme 23)[18] The unprotected N-H ketimines alleviate problems caused by E/Z isomerism in the asymmetric catalytic reactions, although they generally have lower stability. Morimoto et al. showed that the unprotected N-H ketimines are able to react with malonates, esters, oxindoles, and cyclic beta-keto-nitiles under different temperatures to give products whose diastereoselectivities can be controlled under asymmetric catalysis.

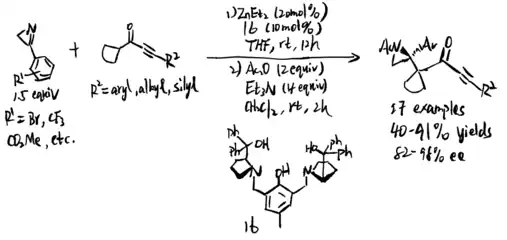
In 2018, Zn-ProPhenol was used to catalyze asymmetric Mannich reaction of butenolides with perfluoroalkyl alkynyl ketimines, which is proposed by Trost et al. (scheme 24)[19] Several features about this reaction were reported: 1. Ketimines are more electrophilic, resulted by fluorinated groups 2. Alkyne group has less steric effect, and its high s character makes ketimines more electrophilic 3. Steric difference between alkynyl and perfluoroalkyl groups would give ketimines in a single diastereomeric form, which is critical for high stereoselectivities. The reaction is broad in scope, which allows further contribution in medicinal field and synthetic chemistry.

Chiral amines like amino acid derivatives can catalyze many enantioselective transformations, although enantiomers of amino acids are hard to access sometimes. In 2019, Lan et al. proposed a strategy to apply their chiral amine catalyst with minimal modification to the Mannich reaction of alkynyl ketiminoesters. (scheme 25)[20] Their application shows that catalyst 26 and MeCN can be replaced by 28 and dichloroethane for many alkynyl trifluoromethyl-, pyrazolinone-derived-, and isatin-derived ketimines to give the same stereoselectivities and yields.
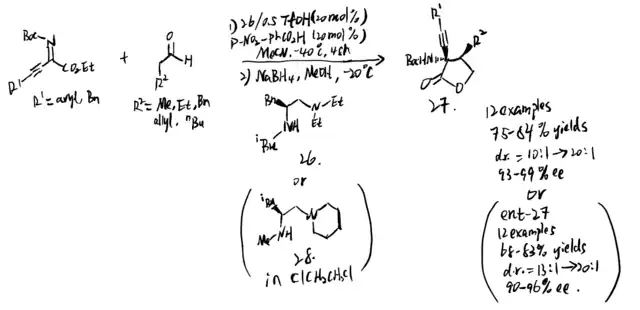
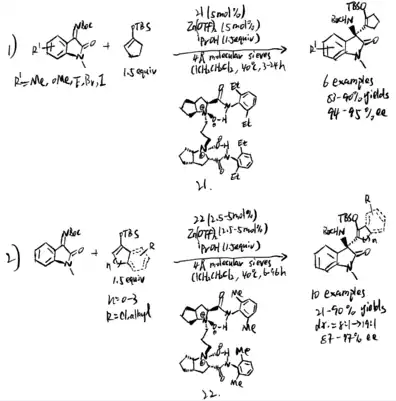
It is hard to control E/Z selectivity for ketimines having two structurally similar substituents, which makes such kind of ketimines merely used in asymmetric catalysis field comparing to ketimines that have single diastereomeric form. In 2021, Kano et al. proposed method to use alkynyl ketimines as synthetic equivalents of dialkyl ketimines, and dialkyl ketimes are a typical example of isomeric ketimine mixtures. The alkynyl groups can be reduced by hydrogenation back to corresponding alkyl groups. Moreover, a single diastereomeric alkynyl alkyl N-protected ketimines can be obtained by using steric difference between alkyl and alkynyl group, which makes them be stereoselectively controlled by chiral catalyst to give asymmetric Mannich product. Additionally, small steric hindrance and high s character of alkynyl group result in its high nucleophilicity, making alkynyl-substituted ketimines more reactive than alkyl counterparts. Even though additional step of hydrogenation is needed, the advantages provided by alkynyl group are still impressive. To solve the problem that a single diastereomeric form of N-Boc alkyl alkynyl ketimines are difficult to access, Kano et al. proposed a synthesis to form these ketimines (reaction 1 and 2), followed by operating chiral amine-catalyzed Mannich reactions (reaction 3). (scheme 26)[21] The chiral amine catalyzed the asymmetric Mannich reaction of aldehyde and (Z)-alkyl alkynyl ketimines. The method they used is stereodivergent, in which the phenylcyclopropane amine 29 gives syn products, and the proline catalyst gives anti product. After the hydrogenation step, they successfully obtained chiral amines substituted with two similar alkyl groups, which is hard to obtained in a single enantiomeric compound.

Applications
Ketimine Mannich catalysis has been identified as a synthetic tool for primary alcohols.[2] A useful path to dihydroquinazolinone, a precursor to drugs with anti-viral and cardiovascular benefits, was also found by Liu[22] et Al. using N-Heterocyclic carbene-catalyzation [4+2] annulation of β-methyl enals.
References
- ↑ Bjørgo, Johannes; Boyd, Derek R.; Watson, Christopher G.; Jennings, W. Brian (1974). "Equilibrium distribution of E–Z-ketimine isomers". J. Chem. Soc., Perkin Trans. 2 (7): 757–762. doi:10.1039/p29740000757. ISSN 0300-9580.
- 1 2 3 4 List, Benjamin; Pojarliev, Peter; Biller, William T.; Martin, Harry J. (2010-05-20). "ChemInform Abstract: The Proline-Catalyzed Direct Asymmetric Three-Component Mannich Reaction: Scope, Optimization, and Application to the Highly Enantioselective Synthesis of 1,2-Amino Alcohols". ChemInform. 33 (29): no. doi:10.1002/chin.200229039. ISSN 0931-7597.
- ↑ Ishitani, Haruro; Ueno, Masaharu; Kobayashi, Shū (1997-07-01). "Catalytic Enantioselective Mannich-Type Reactions Using a Novel Chiral Zirconium Catalyst". Journal of the American Chemical Society. 119 (30): 7153–7154. doi:10.1021/ja970498d. ISSN 0002-7863.
- ↑ Hajos, Zoltan G.; Parrish, David R. (June 1974). "Asymmetric synthesis of bicyclic intermediates of natural product chemistry". The Journal of Organic Chemistry. 39 (12): 1615–1621. doi:10.1021/jo00925a003. ISSN 0022-3263.
- ↑ Kano, Taichi; Aota, Yusuke; Maruoka, Keiji (2017-11-27). "Asymmetric Synthesis of Less Accessible α-Tertiary Amines from Alkynyl Z-Ketimines". Angewandte Chemie International Edition. 56 (51): 16293–16296. doi:10.1002/anie.201710084. ISSN 1433-7851. PMID 29110376.
- ↑ Reddy, K. Nagarjuna; Rao, M. V. Krishna; Sridhar, B.; Subba Reddy, B. V. (2019-08-07). "BINOL Phosphoric Acid‐Catalyzed Asymmetric Mannich Reaction of Cyclic N‐Acyl Ketimines with Cyclic Enones". Chemistry: An Asian Journal. 14 (17): 2958–2965. doi:10.1002/asia.201900556. ISSN 1861-4728. PMID 31241858. S2CID 195658849.
- ↑ Wang, You-Qing; Zhang, Yongna; Pan, Kun; You, Junxiong; Zhao, Jin (2013-11-13). "Direct Organocatalytic Asymmetric Mannich Addition of 3-Substituted-2H-1,4-Benzoxazines: Access to Tetrasubstituted Carbon Stereocenters". Advanced Synthesis & Catalysis. 355 (17): 3381–3386. doi:10.1002/adsc.201300654. ISSN 1615-4150.
- ↑ Wu, Ling-Ling; Xiang, Yang; Yang, Da-Cheng; Guan, Zhi; He, Yan-Hong (2016). "Biocatalytic asymmetric Mannich reaction of ketimines using wheat germ lipase". Catalysis Science & Technology. 6 (11): 3963–3970. doi:10.1039/c5cy01923k. ISSN 2044-4753.
- ↑ Hu, Haipeng; Xu, Jinxiu; Liu, Wen; Dong, Shunxi; Lin, Lili; Feng, Xiaoming (2018-09-05). "Copper-Catalyzed Asymmetric Addition of Tertiary Carbon Nucleophiles to 2H-Azirines: Access to Chiral Aziridines with Vicinal Tetrasubstituted Stereocenters". Organic Letters. 20 (18): 5601–5605. doi:10.1021/acs.orglett.8b02274. ISSN 1523-7060. PMID 30183317. S2CID 52159388.
- ↑ Zhang, Hai-Jun; Xie, Yan-Cheng; Yin, Liang (2019-04-12). "Copper(I)-catalyzed asymmetric decarboxylative Mannich reaction enabled by acidic activation of 2H-azirines". Nature Communications. 10 (1): 1699. Bibcode:2019NatCo..10.1699Z. doi:10.1038/s41467-019-09750-5. ISSN 2041-1723. PMC 6461707. PMID 30979892.
- ↑ Trost, Barry M.; Zhu, Chuanle (2020-12-03). "Zn-ProPhenol Catalyzed Enantioselective Mannich Reaction of 2H-Azirines with Alkynyl Ketones". Organic Letters. 22 (24): 9683–9687. doi:10.1021/acs.orglett.0c03737. ISSN 1523-7060. PMID 33269592. S2CID 227258883.
- ↑ Rong, Meng-Yu; Li, Jin-Shan; Zhou, Yin; Zhang, Fa-Guang; Ma, Jun-An (2020-12-31). "Correction to "Catalytic Enantioselective Synthesis of Difluoromethylated Tetrasubstituted Stereocenters in Isoindolones Enabled by a Multiple-Fluorine System"". Organic Letters. 23 (2): 623. doi:10.1021/acs.orglett.0c04131. ISSN 1523-7060. PMID 33382266. S2CID 229930648.
- ↑ Sawa, Masanao; Miyazaki, Shotaro; Yonesaki, Ryohei; Morimoto, Hiroyuki; Ohshima, Takashi (2018-08-14). "Catalytic Enantioselective Decarboxylative Mannich-Type Reaction of N-Unprotected Isatin-Derived Ketimines". Organic Letters. 20 (17): 5393–5397. doi:10.1021/acs.orglett.8b02306. ISSN 1523-7060. PMID 30106593. S2CID 52007498.
- ↑ Ding, Ransheng; De los Santos, Zeus A.; Wolf, Christian (2019-02-06). "Catalytic Asymmetric Mannich Reaction of α-Fluoronitriles with Ketimines: Enantioselective and Diastereodivergent Construction of Vicinal Tetrasubstituted Stereocenters". ACS Catalysis. 9 (3): 2169–2176. doi:10.1021/acscatal.8b05164. ISSN 2155-5435. PMC 6449049. PMID 30956891.
- ↑ Kang, Tengfei; Hou, Liuzhen; Ruan, Sai; Cao, Weidi; Liu, Xiaohua; Feng, Xiaoming (2020-08-03). "Lewis acid-catalyzed asymmetric reactions of β,γ-unsaturated 2-acyl imidazoles". Nature Communications. 11 (1): 3869. Bibcode:2020NatCo..11.3869K. doi:10.1038/s41467-020-17681-9. ISSN 2041-1723. PMC 7398931. PMID 32747706.
- ↑ Takeda, Tadahiro; Kondoh, Azusa; Terada, Masahiro (2016-03-08). "Construction of Vicinal Quaternary Stereogenic Centers by Enantioselective Direct Mannich-Type Reaction Using a Chiral Bis(guanidino)iminophosphorane Catalyst". Angewandte Chemie International Edition. 55 (15): 4734–4737. doi:10.1002/anie.201601352. ISSN 1433-7851. PMID 26954063.
- ↑ Lin, Shaoquan; Kumagai, Naoya; Shibasaki, Masakatsu (2016-02-02). "Direct Catalytic Asymmetric Mannich-type Reaction of α,β-Unsaturated γ-Butyrolactam with Ketimines". Chemistry: A European Journal. 22 (10): 3296–3299. doi:10.1002/chem.201600034. ISSN 0947-6539. PMID 26834086.
- ↑ Sawa, Masanao; Morisaki, Kazuhiro; Kondo, Yuta; Morimoto, Hiroyuki; Ohshima, Takashi (2017-09-26). "Direct Access to N-Unprotected α- and/or β-Tetrasubstituted Amino Acid Esters via Direct Catalytic Mannich-Type Reactions Using N-Unprotected Trifluoromethyl Ketimines". Chemistry: A European Journal. 23 (67): 17022–17028. doi:10.1002/chem.201703516. ISSN 0947-6539. PMID 28950035.
- ↑ Trost, Barry M.; Hung, Chao‐I. (Joey); Scharf, Manuel J. (2018-07-26). "Direct Catalytic Asymmetric Vinylogous Additions of α,β‐ and β,γ‐Butenolides to Polyfluorinated Alkynyl Ketimines". Angewandte Chemie International Edition. 57 (35): 11408–11412. doi:10.1002/anie.201806249. ISSN 1433-7851. PMID 29969528. S2CID 49677124.
- ↑ Dai, Jun; Wang, Zhuang; Deng, Yuhua; Zhu, Lei; Peng, Fangzhi; Lan, Yu; Shao, Zhihui (2019-11-15). "Enantiodivergence by minimal modification of an acyclic chiral secondary aminocatalyst". Nature Communications. 10 (1): 5182. Bibcode:2019NatCo..10.5182D. doi:10.1038/s41467-019-13183-5. ISSN 2041-1723. PMC 6858435. PMID 31729388.
- ↑ Homma, Chihiro; Takeshima, Aika; Kano, Taichi; Maruoka, Keiji (2021). "Construction of chiral α-tert-amine scaffolds via amine-catalyzed asymmetric Mannich reactions of alkyl-substituted ketimines". Chemical Science. 12 (4): 1445–1450. doi:10.1039/d0sc05269h. ISSN 2041-6520. PMC 8179053. PMID 34163907. S2CID 229395873.
- ↑ Kano, Taichi; Song, Sunhwa; Kubota, Yasushi; Maruoka, Keiji (2011-12-15). "Highly Diastereo- and Enantioselective Mannich Reactions of Synthetically Flexible Ketimines with Secondary Amine Organocatalysts". Angewandte Chemie International Edition. 51 (5): 1191–1194. doi:10.1002/anie.201107375. ISSN 1433-7851. PMID 22173941.