Memory allocation is a process that determines which specific synapses and neurons in a neural network will store a given memory.[1][2][3] Although multiple neurons can receive a stimulus, only a subset of the neurons will induce the necessary plasticity for memory encoding. The selection of this subset of neurons is termed neuronal allocation. Similarly, multiple synapses can be activated by a given set of inputs, but specific mechanisms determine which synapses actually go on to encode the memory, and this process is referred to as synaptic allocation. Memory allocation was first discovered in the lateral amygdala by Sheena Josselyn and colleagues in Alcino J. Silva's laboratory.[4]
At the neuronal level, cells with higher levels of excitability (for example lower slow afterhyperpolarization[5]) are more likely to be recruited into a memory trace, and substantial evidence exists implicating the cellular transcription factor CREB (cyclic AMP responsive element-binding protein) in this process.[5][6] Certain synapses on recruited neurons are more likely to undergo an enhancement of synaptic strength (known as Long-term potentiation (LTP))[7] and proposed mechanisms that might contribute to allocation at the synaptic level include synaptic tagging, capture, and synaptic clustering.[3]
Neuronal allocation
Neuronal allocation is a phenomenon that accounts for how specific neurons in a network, and not others that receive similar input, are committed to storing a specific memory.[3]
The role of CREB in neuronal allocation
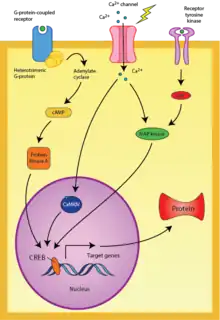
The transcription factor cAMP response element-binding protein (CREB) is a well-studied mechanism of neuronal memory allocation. Most studies to date use the amygdala as a model circuit, and fear-related memory traces in the amygdala are mediated by CREB expression in the individual neurons allocated to those memories.[4][5][8] CREB modulates cellular processes that lead to neuronal allocation, particularly with regards to dendritic spine density and morphology.[9] Many of the memory mechanisms studied to date are conserved across different brain regions, and it is likely that the mechanisms of fear-based memory allocation found in the amygdala will also be similarly present for other types of memories throughout different brain regions.[3] Indeed, Sano and colleagues in the Silva lab showed that CREB also regulates neuronal memory allocation in the amygdala.[10]
CREB may be activated by multiple pathways. For example, the cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) pathways appear to participate in neuronal allocation.[3] When activated by the second messengers such as cAMP and calcium ions, enzymes such as PKA and MAP kinase can translocate to the nucleus and phosphorylate CREB to initiate transcription of target genes.[11][12] PKA inhibitors can block the development of long-lasting LTP, and this is accompanied by a reduction in the transcription of genes modulated by the CREB protein.[13]
Metaplasticity in neuronal allocation
Metaplasticity is a term describing the likelihood that a given stimulus will induce neuronal plasticity, based on the previous activity experienced by that neuron. Several studies provide evidence that neurons receiving “priming activity” (such as neurotransmitters, paracrine signals, or hormones) minutes to days prior will show a lower threshold for induction of long term potentiation (LTP).[14][15][16] Other studies find that activation of NMDARs can also raise the stimulation threshold for induction of LTP.[3] Thus, similar inputs on groups of neurons may induce LTP in some but not others based on prior activity of those neurons.[17]
Signaling mechanisms implicated in these metaplastic effects include autophosphorylation of αCaMKII,[18] changes in NMDA receptor subunit composition,[19] and activation of voltage-dependent calcium channels.[18][20] These metaplastic effects regulate memory destabilization and reconsolidation.[21]
Synaptic allocation
Synaptic allocation pertains to mechanisms that influence how synapses come to store a given memory.[3] Intrinsic to the idea of synaptic allocation is the concept that multiple synapses can be activated by a given set of inputs, but specific mechanisms determine which synapses actually go on the encode the memory. Allocation of memories to specific synapses are key to determining where memories are stored.
Synaptic tagging and capture

Synaptic activity can generate a synaptic tag, which is a marker that allows the stimulated spine to subsequently capture newly transcribed plasticity molecules such as Arc. Synaptic activity can also engage the translation and transcription machinery. Weak stimulation can create synaptic tags but will not engage the translation and transcription machinery, whereas strong stimulation will create synaptic tags and also engage the translation and transcription machinery. Newly generated plasticity-related proteins (PRPs) can be captured by any tagged synapses, but untagged synapses are not eligible to receive new PPs. After a certain time period, synapses will lose their tag and return to their initial state. Furthermore, the supply of new PRPs will deplete. The tags and new PRPs must overlap in time to capture the PRPs.[17][22][23]
The synaptic tag is inversely related to time between inducing stimuli, and is said to be temporarily asymmetrical. Furthermore, the tagging is also inversely related to the distance between spines, an important spatial properties of tagging. Conversely confirming the temporal and spatial properties of the synaptic tagging, subsequent imaging studies revealed that there are not only temporal constraints but also structural constraints that limit synaptic tagging and capture mechanisms. Overall, these studies demonstrate the complexity of synaptic tagging and capture, and give further insight into how exactly this mechanism occurs.[3]
Spine clustering
Synaptic clustering refers to the addition of new spines to a dendritic area where other spines have been added by previous learning.[3] Spine clustering may result in the amplification of synaptic inputs via diffusible molecular crosstalk that occurs near activated spines.1 For example, studies have shown that signaling molecules synthesized at one spine, (e.g. activated RAS and/or RHOA), may diffuse out and influence spine growth at nearby sites.[24] The Rho GTPase CDC42 may also contribute to spine clustering by driving long-term spine volume increases. Recent studies also suggest that this process may be regulated by NMDA receptor activation and nitric oxide stimulation.[3][25]
Spine clustering in the motor cortex reflects a morphological mechanism for synaptic storage of specific motor memories. These clustered spines are more stable than non-clustered new spines. This type of addition of spines occurs in a specific pattern, meaning that spines added after one task will not cluster with spines after an alternative task.[26] Loss of spine clustering is also a possibility as shown in some fear conditioning experiments, leading to the net loss of spines in the frontal association cortex, a region strongly associated in fear conditioning, which strongly correlates with memory on recall. Once spines were added after fear extinction had a similar orientation to the spines lost during the original fear conditioning.[27]
Mechanisms that link memories across time
Denise Cai in Alcino J. Silva's laboratory found that memory allocation mechanisms are used to connect or link memories across time.[28] In their studies they demonstrated that one contextual memory triggers the activation of CREB and subsequent enhancements in excitability in a subset of hippocampal CA1 neurons, such that a subsequent contextual memory, occurring within 5 hours, can be allocated to some of the same CA1 neurons that stored the first contextual memory. As a consequence of this overlap between the CA1 memory engrams for the two contextual memories, recall of one contextual memory activates the retrieval of the second memory. These studies also showed that contextual memory linking mechanisms are disrupted in the aging brain, and that increasing excitability in a subset of CA1 neurons reverses these memory linking deficits. It is very likely that impairments in CREB and neuronal excitability in aging brains could account for abnormalities in memory linking and possibly related source memory problems (source amnesia) associated with aging. In July 2018, in a special issue about "13 Discoveries that Could Change Everything", Scientific American highlighted the Silva laboratory's discovery of Memory Allocation and Linking [29]
Current and future research
Integrating synaptic and neuronal allocation
Experiments have yet to investigate the interaction of allocative mechanisms between the neuronal and synaptic levels. The two classes of processes are very likely to be interconnected considering the relationship between neurons and synapses in a neuronal network. For example, the synaptic tagging and capture involved in synaptic allocation requires the allocation of the neurons to which the synapses belong to. Moreover, increases in neuronal excitability in a given neuronal ensemble may affect some dendrites more than others, thus biasing memory storage to synapses in dendrites with higher excitability.[30][31] Similarly, on the recruited neurons displaying increased excitability, specific synapses need to be selected for in order to store the information in the form of synaptic plasticity.
One aspect of integration involves metaplasticity and how acquisition and storage of one memory changes the neural circuit to affect the storage and properties of a subsequent memory. Cellular excitability has been proposed as one of the mechanisms responsible for heterosynaptic metaplasticity, the modulation of subsequent plasticity at different synapses.[32] CREB functions through elevating cell excitability as described above, thus it is also possibly involved in hetrerosynaptic metaplasticity. Synaptic tagging and capture, as introduced in sections above, can result in a weak memory (capable of triggering only E-LTP), which would otherwise be forgotten, but it can be strengthened and stabilized by a strong memory (capable of triggering L-LTP), which is a form of heterosynaptic plasticity.
Future research
Despite extensive research into the individual mechanisms of memory allocation, there are few studies investigating the integration of these mechanisms. It has been proposed that understanding the implications of the molecular, cellular and systemic mechanisms of these processes may elucidate how they are coordinated and integrated during memory formation.[3] For example, identifying the plasticity-related proteins (PRPs) involved in synaptic tagging and capture as well as the upstream and downstream molecules of CREB can help reveal potential interactions. Investigating the functional significance of these mechanisms will require tools that can directly manipulate and image the processes involved in the proposed mechanisms in vivo.[3] For instance, it is possible that the behavioral interactions ascribed to synaptic tagging and capture are caused by protein synthesis-dependent increases in neuromodulators such as dopamine rather than by synaptic tagging mechanisms. Examining the behavioral effects under direct manipulation can help rule out these other possible causes.
See also
References
- ↑ Won, J. and A.J. Silva, Molecular and cellular mechanisms of memory allocation in neuronetworks. Neurobiol Learn Mem, 2007. PMC 2673809
- ↑ Silva, A.J., Zhou, Y, Rogerson, T, Shobe, J and Balaji, J. Molecular and Cellular Approaches to Memory Allocation in Neural Circuits. Science, Oct 16 2009;326(5951):391-5. PMID 19833959.
- 1 2 3 4 5 6 7 8 9 10 11 12 Rogerson, T. et al. Synaptic tagging during memory allocation. Nature Rev. Neurosci 15, 157-169 (2014)
- 1 2 Han, J. H., Kushner, S. A., Yiu, A. P., Cole, C. J., Matynia, A., Brown, R. A., ... & Josselyn, S. A. (2007). Neuronal competition and selection during memory formation. science, 316(5823), 457-460.
- 1 2 3 Zhou, Y., Won, J., Karlsson, M. G., Zhou, M., Rogerson, T., Balaji, J., ... & Silva, A. J. (2009). CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nature neuroscience, 12(11), 1438-1443.
- ↑ Yiu, A. P. et al. Neurons Are Recruited to a Memory Trace Based on Relative Neuronal Excitability Immediately before Training. Neuron 83, 722-735 (2014)
- ↑ Bliss, T. V., & Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature, 361(6407), 31-39.
- ↑ Han, J. H., Kushner, S. A., Yiu, A. P., Hsiang, H. L. L., Buch, T., Waisman, A., ... & Josselyn, S. A. (2009). Selective erasure of a fear memory. Science, 323(5920), 1492-1496.
- ↑ Sargin, D., Mercaldo, V., Yiu, A. P., Higgs, G., Han, J. H., Frankland, P. W., & Josselyn, S. A. (2013). CREB regulates spine density of lateral amygdala neurons: implications for memory allocation. Frontiers in behavioral neuroscience, 7.
- ↑ Sano, Y, Shobe, JL, Zhou, M, Huang, S, Cai, DJ, Roth, BL, Kamata, M, and Silva, AJ. CREB regulates memory allocation in the insular cortex. Curr Biol. 2014 Nov 13;24(23):2833-283 (2014) PMID 25454591
- ↑ Adams, J. P., & Sweatt, J. D. (2002). Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annual Review of Pharmacology and Toxicology, 42(1), 135-163.
- ↑ Treisman, R. (1996). Regulation of transcription by MAP kinase cascades. Current Opinion in Cell Biology, 8(2), 205-215.
- ↑ Nguyen, P. V., & Woo, N. H. (2003). Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Progress in Neurobiology,71(6), 401-437.
- ↑ Goussakov, I. V., Fink, K., Elger, C. E., & Beck, H. (2000). Metaplasticity of mossy fiber synaptic transmission involves altered release probability. The Journal of Neuroscience, 20(9), 3434-3441.
- ↑ Abraham, W. C., & Tate, W. P. (1997). Metaplasticity: a new vista across the field of synaptic plasticity. Progress in Neurobiology, 52(4), 303-323.
- ↑ Koon, Alex C., et al. "Autoregulatory and paracrine control of synaptic and behavioral plasticity by octopaminergic signaling." Nature neuroscience 14.2 (2011): 190-199.
- 1 2 3 Rudy, J. (2014). Specific mechanisms: Targeting plasticity products. In The neurobiology of Learning and Memory (Second ed., pp. 113-116). Sinauer Associates.
- 1 2 Zhang, L., Kirschstein, T., Sommersberg, B., Merkens, M., Manahan-Vaughan, D., Elgersma, Y., & Beck, H. (2005). Hippocampal synaptic metaplasticity requires inhibitory autophosphorylation of Ca2+/calmodulin-dependent kinase II. The Journal of Neuroscience, 25(33), 7697-7707.
- ↑ Cho, K. K., Khibnik, L., Philpot, B. D., & Bear, M. F. (2009). The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proceedings of the National Academy of Sciences, 106(13), 5377-5382.
- ↑ Lee, M. C., Yasuda, R., & Ehlers, M. D. (2010). Metaplasticity at single glutamatergic synapses. Neuron, 66(6), 859-870.
- ↑ Finnie, Peter SB, and Karim Nader. "The role of metaplasticity mechanisms in regulating memory destabilization and reconsolidation." Neuroscience & Biobehavioral Reviews 36.7 (2012): 1667-1707.
- ↑ Frey, U. & Morris, R. G. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 21, 181–188 (1998)
- ↑ Govindarajan, A. et al. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron 69, 132–146 (2011).
- ↑ Harvey, C. D. et al. The spread of Ras activity triggered by activation of a single dendritic spine. Science 321, 136–140 (2008).
- ↑ Murakoshi, H., Wang, H. & Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472, 100–104 (2011)
- ↑ Fu, M. et al. Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature 483, 92–95 (2012).
- ↑ Lai, C. S. et al. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature 483, 87–91 (2012).
- ↑ Cai DJ, Aharoni D, Shuman T, Shobe J, Biane J, Song W, Wei B, Veshkini M, La-Vu M, Lou J, Flores S, Kim I, Sano Y, Zhou M, Baumgaertel K, Lavi A, Kamata M, Tuszynski M, Mayford M, Golshani P, Silva AJ. A shared neural ensemble links distinct contextual memories encoded close in time. Nature 2016 May 23;534(7605):115-8
- ↑ Silva, AJ How one memory attaches to another. In Revolutions in Science: Discoveries that could change everything. Scientific American; July 2018, Volume 27, Issue 3s
- ↑ Larkum, M. E. & Nevian, T. Synaptic clustering by dendritic signalling mechanisms. Curr. Opin. Neurobiol. 18, 321–331 (2008)
- ↑ Losonczy, A., Makara, J. K. & Magee, J. C. Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441 (2008)
- ↑ Frick, A. & Johnston, D. Plasticity of dendritic excitability. J. Neurobiol. 64, 100–115 (2005)
| Basic science |
|  |
|---|---|---|
| Clinical neuroscience |
| |
| Cognitive neuroscience | ||
| Interdisciplinary fields |
| |
| Concepts |
| |
| ||