Proximity ligation-assisted chromatin immunoprecipitation sequencing (PLAC-seq) is a chromatin conformation capture(3C)-based technique to detect and quantify genomic chromatin structure from a protein-centric approach.[1] PLAC-seq combines in situ Hi-C and chromatin immunoprecipitation (ChIP), which allows for the identification of long-range chromatin interactions at a high resolution with low sequencing costs.[1] Mapping long-range 3-dimensional(3D) chromatin interactions is important in identifying transcription enhancers and non-coding variants that can be linked to human diseases.[2]
Different 3C-based techniques have been used to study the higher-order 3D chromatin structure, and it has been combined with high-throughput sequencing to determine the chromatin structure on a genome-wide level.[3] Hi-C is one of the most widely used 3C-based techniques because it allows for high-resolution (kilobase-scale) genome-topology identification. However, it requires billions of sequencing reads which has limited its application.[2] Another commonly used 3C-based technique is chromatin interaction analysis by paired-end tag sequencing (ChiA-PET).[2] ChiA-PET can identify long-range interactions of transcription promoters and enhancers at a high resolution but requires millions of cells.[2]
PLAC-seq alleviates these issues by using in situ Hi-C, which creates long-range DNA contacts in situ in the nucleus before lysis.[3] Unlike ChiA-PET which performs ChIP and proximity ligation after chromatin shearing, performing proximity ligation in the nuclei first prevents large disruptions of protein/DNA complexes.[2] This decreases false-positive interactions and improves DNA contact capture efficiency, meaning that PLAC-seq is more accurate and requires fewer cells.[1]
History
PLAC-seq was developed in 2016[2] and an almost identical technique called HiChIP was also developed in the same year.[3] Both methods combine in situ Hi-C and ChIP but have different library preparation methods.[1] While PLAC-seq uses biotin pull-down followed by end-repair, adapter ligation, and PCR, HiChIP usesTn5 tagmentation, biotin pull-down, and PCR.[1] However, both techniques can use the same quality control and data analysis techniques.[1]
Different computation software tools can be used to analyze the data from PLAC-seq, for example, Fit-Hi-C,[4] HiCCUPS,[5] Mango,[6] Hichipper,[7] MAPS,[8] and FitHiChIP.[9] Many of the earlier software tools were developed for other 3C-based technologies and were not optimized for PLAC-seq/HiChIP data. Fit-Hi-C and HiCCUPS, both developed in 2014, were mainly developed for Hi-C data, and utilize a matrix-balancing-based normalization approach.[4][5] Mango was developed in 2015, and is mainly used for ChIA-PET data, but has high false-positive rates in analyzing PLAC-seq/HiChIP data due to the different biases.[6][8] Hichipper was developed in 2018 to alleviate this issue and introduced a bias-correcting algorithm, but it still has difficulties identifying protein interactions between protein binding and non-protein binding regions on the chromosome.[7][8] MAPS and FitHiChIP were developed in 2019 as a PLAC-seq/HiChIP-specific analysis pipeline, and are generally thought to be more effective than the existing models to analyze PLAC-seq/HiChIp data.[8][9]
Procedure
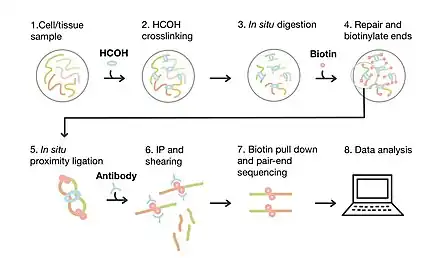
The general workflow of PLAC-seq involves cell harvesting and crosslinking, in situ digestion and proximity ligation, ChIP, library construction, sequencing, and data analysis. The first step of PLAC-seq includes the preparation and crosslinking of cell and tissue samples, which typically begins with cell collection through centrifugation. The next step involves the use of a DNA crosslinking agent such as formaldehyde (HCOH) followed by the addition of glycine to stop the crosslinking reaction. The cross-linked cells can then be pelleted by centrifugation and either stored at -80 or used in the next step of the procedure. In situ digestion involves cell lysis with the use of a lysis buffer followed by digestion with a restriction enzyme MboI. This step allows for uniform digestion of genetic material while keeping the crosslinked regions of the chromosome intact. After inactivation of the digestion reaction, dNTPs and biotin are added in order to repair overhangs and mark the DNA for pull down respectively. In situ proximity ligation occurs when the biotinylated ends of the crosslinked DNA are ligated with each other. Chromatin fragmentation by sonication allows for the shearing of non-crosslinked fragments of DNA. This is followed by immunoprecipitation of biotinylated DNA through the use of antibody-coated beads. The DNA is then reverse-crosslinked and purified using column-based DNA purification or phenol-chloroform extraction. The library construction step first involves the pull-down of biotinylated DNA and the addition of sequencing adapters. The cycle number for amplification needs to be determined prior to the final amplification and library purification. Data analysis of PLAC-seq sequencing data can be carried out in multiple ways, however, the common methods involve the use of Fit-Hi-C,[4] FitHiChIP,[9] and MAPS.[8] Data analysis involves mapping to a reference genome, using software tools such as Hichipper[7] to identify peaks, and downstream analysis involving peak comparison and functional enrichment analysis.[8] The resulting data can also be integrated with other genomic data such as Hi-C or RNA-seq in order to identify potential regulatory networks.
Applications
PLAC-seq was developed to map and analyze long-range chromatin interactions. These interactions have important implications when it comes to the transcriptional regulation of genes.[10]
One challenge for mammalian cells is fitting around two meters of genetic material into a nucleus that is around a few microns in diameter, and at the same time organizing the genetic material to be able to access and use the genetic and epigenetic information. To do this, DNA is compacted around histone octamers into 2D structures, and then further packaged into 3D compartments by various mechanisms such as cis-regulatory interactions and repressive interactions. Therefore, chromosomal regions distant in 2D may have intra- and interchromosomal long-range interactions in 3D. These 3D structures are involved in the induction and repression of genes that have biological implications on basic cell functions such as cell cycle, replication, and development. Aberrant 3D structures have roles in the development of diseases and abnormalities such as cancer.[11] This can involve interactions between promoters and terminators/enhancers through the formation of long-range chromatin loops.[12][13]
PLAC-seq has been utilized to study H3K4me3 and H3K27ac PLACE (PLAC-Enriched) interactions.[2] It has also been used to call for significant H3K4me3-mediated chromatin interactions, thereby allowing for the identification of differential epigenetic modification in different cell types such as those found in the developing human cortex.[14]
Use
Advantages: Compared to ChIA-PET, PLAC-seq requires significantly less amount of starting biological material.[1] With shearing being one of the first steps in ChIA-PET, this leads to the disruption of protein and DNA complexes. PLAC-seq avoids this by having the crosslinking reaction precede the shearing process. Furthermore, PLAC-seq requires fewer sequencing reads than Hi-C.[1] While ChIA-PET requires 100 million starting cells, PLAC-seq only requires 5 million cells.[2] Even with 20-fold fewer cells, PLAC-seq was able to produce more reads (175 million) with a fewer PCR duplication rate (33%) than ChIA-PET (16 million, and 44% respectively).[2] PLAC-seq was also nearly 100 times more cost-effective than ChIA-PET.[1]
Disadvantages: While many of the 3C-based techniques have different biases from the protocols, PLAC-seq (and HiChIP) data have biases from immunoprecipitation efficiencies that need to be corrected for in the computational step.[15] Effective ways of reducing and/or removing the different biases in 3C-based technologies is still being studied.[15]
References
- 1 2 3 4 5 6 7 8 9 Yu, Miao; Juric, Ivan; Abnousi, Armen; Hu, Ming; Ren, Bing (2021). "Proximity Ligation-Assisted ChIP-Seq (PLAC-Seq)". Enhancers and Promoters: Methods and Protocols. Methods in Molecular Biology. Vol. 2351. New York, NY: Springer US. pp. 181–199. doi:10.1007/978-1-0716-1597-3_10. ISBN 978-1-07-161597-3. PMID 34382190. S2CID 236988360. Retrieved 2023-02-27.
- 1 2 3 4 5 6 7 8 9 Fang, Rongxin; Yu, Miao; Li, Guoqiang; Chee, Sora; Liu, Tristin; Schmitt, Anthony D; Ren, Bing (December 2016). "Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq". Cell Research. 26 (12): 1345–1348. doi:10.1038/cr.2016.137. ISSN 1001-0602. PMC 5143423. PMID 27886167.
- 1 2 3 Mumbach, Maxwell R.; Rubin, Adam J.; Flynn, Ryan A.; Dai, Chao; Khavari, Paul A.; Greenleaf, William J.; Chang, Howard Y. (November 2016). "HiChIP: efficient and sensitive analyss of protein-directed genome architecture". Nature Methods. 13 (11): 919–922. doi:10.1038/nmeth.3999. ISSN 1548-7105. PMC 5501173. PMID 27643841.
- 1 2 3 Ay, Ferhat; Bailey, Timothy L.; Noble, William Stafford (June 2014). "Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts". Genome Research. 24 (6): 999–1011. doi:10.1101/gr.160374.113. ISSN 1549-5469. PMC 4032863. PMID 24501021.
- 1 2 Rao, Suhas S. P.; Huntley, Miriam H.; Durand, Neva C.; Stamenova, Elena K.; Bochkov, Ivan D.; Robinson, James T.; Sanborn, Adrian L.; Machol, Ido; Omer, Arina D.; Lander, Eric S.; Aiden, Erez Lieberman (2014-12-18). "A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping". Cell. 159 (7): 1665–1680. doi:10.1016/j.cell.2014.11.021. ISSN 1097-4172. PMC 5635824. PMID 25497547.
- 1 2 Phanstiel, Douglas H.; Boyle, Alan P.; Heidari, Nastaran; Snyder, Michael P. (2015-10-01). "Mango: a bias-correcting ChIA-PET analysis pipeline". Bioinformatics. 31 (19): 3092–3098. doi:10.1093/bioinformatics/btv336. ISSN 1367-4811. PMC 4592333. PMID 26034063.
- 1 2 3 Lareau, Caleb A.; Aryee, Martin J. (2018-02-28). "hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data". Nature Methods. 15 (3): 155–156. doi:10.1038/nmeth.4583. ISSN 1548-7105. PMC 10572103. PMID 29489746. S2CID 3569124.
- 1 2 3 4 5 6 Juric, Ivan; Yu, Miao; Abnousi, Armen; Raviram, Ramya; Fang, Rongxin; Zhao, Yuan; Zhang, Yanxiao; Qiu, Yunjiang; Yang, Yuchen; Li, Yun; Ren, Bing; Hu, Ming (April 2019). "MAPS: Model-based analysis of long-range chromatin interactions from PLAC-seq and HiChIP experiments". PLOS Computational Biology. 15 (4): –1006982. Bibcode:2019PLSCB..15E6982J. doi:10.1371/journal.pcbi.1006982. ISSN 1553-7358. PMC 6483256. PMID 30986246.
- 1 2 3 Bhattacharyya, Sourya; Chandra, Vivek; Vijayanand, Pandurangan; Ay, Ferhat (2019-09-17). "Identification of significant chromatin contacts from HiChIP data by FitHiChIP". Nature Communications. 10 (1): 4221. Bibcode:2019NatCo..10.4221B. doi:10.1038/s41467-019-11950-y. ISSN 2041-1723. PMC 6748947. PMID 31530818.
- ↑ Li, En; Liu, Han; Huang, Liangliang; Zhang, Xiangbo; Dong, Xiaomei; Song, Weibin; Zhao, Haiming; Lai, Jinsheng (2019-06-14). "Long-range interactions between proximal and distal regulatory regions in maize". Nature Communications. 10 (1): 2633. Bibcode:2019NatCo..10.2633L. doi:10.1038/s41467-019-10603-4. ISSN 2041-1723. PMC 6572780. PMID 31201330. S2CID 256634176.
- ↑ Deng, Siwei; Feng, Yuliang; Pauklin, Siim (2022-05-04). "3D chromatin architecture and transcription regulation in cancer". Journal of Hematology & Oncology. 15 (1): 49. doi:10.1186/s13045-022-01271-x. ISSN 1756-8722. PMC 9069733. PMID 35509102. S2CID 248508591.
- ↑ Dean, Ann (2011-01-01). "In the loop: long range chromatin interactions and gene regulation". Briefings in Functional Genomics. 10 (1): 3–10. doi:10.1093/bfgp/elq033. ISSN 2041-2649. PMC 3040559. PMID 21258045. Retrieved 2023-02-28.
- ↑ Dekker, Job; Misteli, Tom (October 2015). "Long-Range Chromatin Interactions". Cold Spring Harbor Perspectives in Biology. 7 (10): –019356. doi:10.1101/cshperspect.a019356. ISSN 1943-0264. PMC 4588061. PMID 26430217.
- ↑ Song, Michael; Pebworth, Mark-Phillip; Yang, Xiaoyu; Abnousi, Armen; Fan, Changxu; Wen, Jia; Rosen, Jonathan D.; Choudhary, Mayank N. K.; Cui, Xiekui; Jones, Ian R.; Bergenholtz, Seth; Eze, Ugomma C.; Juric, Ivan; Li, Bingkun; Maliskova, Lenka; Lee, Jerry; Liu, Weifang; Pollen, Alex A.; Li, Yun; Wang, Ting; Hu, Ming; Kriegstein, Arnold R.; Shen, Yin (November 2020). "Cell-type-specific 3D epigenomes in the developing human cortex". Nature. 587 (7835): 644–649. Bibcode:2020Natur.587..644S. doi:10.1038/s41586-020-2825-4. ISSN 1476-4687. PMC 7704572. PMID 33057195.
- 1 2 Zhong, Wujuan; Liu, Weifang; Chen, Jiawen; Sun, Quan; Hu, Ming; Li, Yun (2022-08-19). "Understanding the function of regulatory DNA interactions in the interpretation of non-coding GWAS variants". Frontiers in Cell and Developmental Biology. 10: 957292. doi:10.3389/fcell.2022.957292. ISSN 2296-634X. PMC 9437546. PMID 36060805.