Pathway is the term from molecular biology for a curated schematic representation of a well characterized segment of the molecular physiological machinery, such as a metabolic pathway describing an enzymatic process within a cell or tissue or a signaling pathway model representing a regulatory process that might, in its turn, enable a metabolic or another regulatory process downstream. A typical pathway model starts with an extracellular signaling molecule that activates a specific receptor, thus triggering a chain of molecular interactions.[2] A pathway is most often represented as a relatively small graph with gene, protein, and/or small molecule nodes connected by edges of known functional relations. While a simpler pathway might appear as a chain,[3] complex pathway topologies with loops and alternative routes are much more common. Computational analyses employ special formats of pathway representation.[4][5] In the simplest form, however, a pathway might be represented as a list of member molecules with order and relations unspecified. Such a representation, generally called Functional Gene Set (FGS), can also refer to other functionally characterised groups such as protein families, Gene Ontology (GO) and Disease Ontology (DO) terms etc. In bioinformatics, methods of pathway analysis might be used to identify key genes/ proteins within a previously known pathway in relation to a particular experiment / pathological condition or building a pathway de novo from proteins that have been identified as key affected elements. By examining changes in e.g. gene expression in a pathway, its biological activity can be explored. However most frequently, pathway analysis refers to a method of initial characterization and interpretation of an experimental (or pathological) condition that was studied with omics tools or genome-wide association study.[6] Such studies might identify long lists of altered genes. A visual inspection is then challenging and the information is hard to summarize, since the altered genes map to a broad range of pathways, processes, and molecular functions (with a large gene fraction lacking any annotation). In such situations, the most productive way of exploring the list is to identify enrichment of specific FGSs in it. The general approach of enrichment analyses is to identify FGSs, members of which were most frequently or most strongly altered in the given condition, in comparison to a gene set sampled by chance. In other words, enrichment can map canonical prior knowledge structured in the form of FGSs to the condition represented by altered genes.
Use
The data for pathway analysis come from high throughput biology. This includes high throughput sequencing data and microarray data. Before pathway analysis can be done, each gene's alteration should be evaluated using the omics dataset in either quantitative (differential expression analysis) or qualitative (detection of somatic point mutations or mapping neighbor genes to a disease-associated SNP). It is also possible to combine datasets from different research groups or multiple omics platform with a meta-analysis and cross-platform regularization.[7][8] Further, a list where gene identifiers are accompanied by the alteration attributes is subjected to a pathway analysis. By using pathway analysis software, researchers can determine which FGSs are enriched with the altered experimental genes[9][10] For example, pathway analysis of several independent microarray experiments (meta-analysis) helped to discover potential biomarkers in a single pathway important for fast-to-slow switch fiber type transition in Duchenne muscular dystrophy.[11] In another study meta-analysis identified two biomarkers in blood of patients with Parkinson's disease, which can be useful for monitoring the disease.[12] Candidate gene alleles causative of Alzheimer's disease and elderly dementia where first discovered via genome-wide association study and further validated with network enrichment analysis against FGS consisting of known Alzheimer's genes.[13][14]
Databases
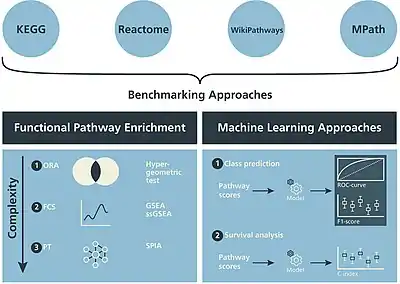
Pathway collections and interaction networks constitute the knowledge base required for a pathway analysis. Pathway content, structure, format, and functionality vary between different database resources such as KEGG,[15] WikiPathways, or Reactome.[16] Also exist proprietary pathways collections used by e.g. Pathway Studio[17] and Ingenuity Pathway Analysis[18] tools. Public online tools can provide pre-compiled and ready-to-go menus of pathways and networks from different open sources (e.g. EviNet).
Methods and software
Pathway analysis software can be found in the form of desktop programs, web-based applications, or packages coded in such languages as R and Python and shared openly through the BioConductor[19] and GitHub[20] projects. The methodology of pathway analysis evolves fast and the classification is still discussable,[21][22] with the following main categories of pathway enrichment analysis applicable to high-throughput data:[21]
Over-representation analysis (ORA)
This method measures the overlap between, on the one hand, a set of genes (or proteins) in an FGS and, on the other hand, a list of most altered genes generally called Altered Gene Sets (AGS). A typical AGS example is a list of top N differentially expressed genes from an RNA-Seq assay. The basic assumption behind ORA is that a biologically relevant pathway can be identified by excess of AGS genes in it compared to the number expected by chance. The aim of ORA is to identify such enriched pathways, judging by statistical significance of the overlap between FGS and AGS as determined either by an appropriate statistic, such as Jaccard index or by a statistical test producing p-values (Fisher's exact test or the test using hypergeometric distribution).
Functional class scoring (FCS)
This method identifies FGS by considering their relative positions in the full list of genes studied in the experiment. This full list should be therefore ranked in advance by a statistic (such as mRNA expression fold-change, Student's t-test etc.) or a p-value - while watching the direction of fold change, since p-values are non-directional. Thus FCS takes into account every FGS gene regardless of its statistical significance and does not require pre-compiled AGS. One of the first and most popular methods deploying the FCS approach was the Gene Set Enrichment Analysis (GSEA).[10]
Pathway topology analysis (PTA)
Similarly to FCS, PTA accounts for high-throughput data for every FGS gene.[23] In addition, specific topological information is used about role, position, and interaction directions of the pathway genes. This requires additional input data from a pathway database in a pre-specified format, such as KEGG Markup Language (KGML). Using this information, PTA estimates a pathway significance by considering how much each individual gene alteration might have affected the whole pathway. Multiple alteration types can be used in parallel (somatic copy-number variations, point mutations etc.) when available.[21] The set of PTA methods includes the Impact Analysis,[24][25] EnrichNet,[26] GGEA,[27] and TopoGSA.[28]
Network enrichment analysis (NEA)
Network enrichment analysis (NEA) has been an extension of gene-set enrichment analysis to the domain of global gene networks[29][30][31][32] The major principle of NEA can be understood in comparison with ORA, where enrichment of FGS in genes of the AGS is determined by how many genes are directly shared by AGS and FGS. In NEA, on the contrary, the global network is searched for network edges that connect any genes of AGS with any genes of FGS. Since enrichment significance is influenced by the highly variable node degrees of individual AGS and FGS genes, it should be determined by a dedicated statistical test, which compares the observed number of network edges to the number expected by chance in the same network context. Some valuable properties of NEA are that:
Commercial solutions
Beyond open-source tools, such as STRING or Cytoscape, a number of companies sell licensed software products to analyse gene sets. While most of the publicly available solutions use online and public pathway collections, the commercial products mostly promote own, proprietary pathways and networks. The choice of such products might be driven by customers' skills, financial and time resources, and needs.[6] Ingenuity, for example, maintains a knowledge base for comparative analysis of gene expression data.[36] Pathways Studio[37] is commercial software which allows searching for biologically relevant facts, analyze experiments, and create pathways. Pathways Studio Viewer[38] is a free resource from the same company for presenting the Pathway Studio interactive pathway collection and database. Two commercial solutions offer PTA: iPathwayGuide from Advaita Corporation and MetaCore from Thomson Reuters.[39] Advaita uses the peer reviewed Impact Analysis method[24][25] while the MetaCore method is unpublished.[39]
Limitations
Lack of annotations
Application of pathway analysis methods depends on annotations found in existing databases, such as gene set membership in pathways, pathway topology, presence of genes in the global network etc. These annotations, however, are far from being complete and have highly variable degrees of confidence. In addition, such information is usually general, i.e. deprived of e.g. cell type, compartment, or developmental context. Therefore, interpretation of pathway analysis results for omics datasets should be done with caution[22] Partially, the problem can be addressed by analysing larger gene sets in a more, such as big pathway collections or global interaction networks.[40]
See also
References
- ↑ Mubeen, Sarah; Hoyt, Charles Tapley; Gemünd, André; Hofmann-Apitius, Martin; Fröhlich, Holger; Domingo-Fernández, Daniel (2019). "The Impact of Pathway Database Choice on Statistical Enrichment Analysis and Predictive Modeling". Frontiers in Genetics. 10: 1203. doi:10.3389/fgene.2019.01203. ISSN 1664-8021. PMC 6883970. PMID 31824580.
- ↑ Berg J. M., Tymoczko J. L., Stryer L. Biochemistry, 5th edition, New York: W. H. Freeman; 2002
- ↑ Ohlrogge J, Browse J (July 1995). "Lipid biosynthesis". The Plant Cell. 7 (7): 957–70. doi:10.1105/tpc.7.7.957. PMC 160893. PMID 7640528. S2CID 219201001.
- ↑ "Main Page - SBML.caltech.edu". sbml.org.
- ↑ "KGML (KEGG Markup Language)". www.genome.jp.
- 1 2 García-Campos MA, Espinal-Enríquez J, Hernández-Lemus E (2015). "Pathway Analysis: State of the Art". Frontiers in Physiology. 6: 383. doi:10.3389/fphys.2015.00383. PMC 4681784. PMID 26733877.
- ↑ Walsh CJ, Hu P, Batt J, Santos CC (August 2015). "Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery". Microarrays. 4 (3): 389–406. doi:10.3390/microarrays4030389. PMC 4996376. PMID 27600230.
- 1 2 Suo C, Hrydziuszko O, Lee D, Pramana S, Saputra D, Joshi H, et al. (August 2015). "Integration of somatic mutation, expression and functional data reveals potential driver genes predictive of breast cancer survival". Bioinformatics. 31 (16): 2607–13. doi:10.1093/bioinformatics/btv164. PMID 25810432.
- ↑ Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM (July 1999). "Systematic determination of genetic network architecture". Nature Genetics. 22 (3): 281–5. doi:10.1038/10343. PMID 10391217. S2CID 14688842.
- 1 2 Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. (October 2005). "Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles". Proceedings of the National Academy of Sciences of the United States of America. 102 (43): 15545–50. Bibcode:2005PNAS..10215545S. doi:10.1073/pnas.0506580102. PMC 1239896. PMID 16199517.
- ↑ Kotelnikova E, Shkrob MA, Pyatnitskiy MA, Ferlini A, Daraselia N (February 2012). "Novel approach to meta-analysis of microarray datasets reveals muscle remodeling-related drug targets and biomarkers in Duchenne muscular dystrophy". PLOS Computational Biology. 8 (2): e1002365. Bibcode:2012PLSCB...8E2365K. doi:10.1371/journal.pcbi.1002365. PMC 3271016. PMID 22319435.
- ↑ Santiago JA, Potashkin JA (February 2015). "Network-based metaanalysis identifies HNF4A and PTBP1 as longitudinally dynamic biomarkers for Parkinson's disease". Proceedings of the National Academy of Sciences of the United States of America. 112 (7): 2257–62. Bibcode:2015PNAS..112.2257S. doi:10.1073/pnas.1423573112. PMC 4343174. PMID 25646437.
- ↑ Reynolds CA, Hong MG, Eriksson UK, Blennow K, Wiklund F, Johansson B, et al. (May 2010). "Analysis of lipid pathway genes indicates association of sequence variation near SREBF1/TOM1L2/ATPAF2 with dementia risk". Human Molecular Genetics. 19 (10): 2068–78. doi:10.1093/hmg/ddq079. PMC 2860895. PMID 20167577.
- ↑ Bennet AM, Reynolds CA, Eriksson UK, Hong MG, Blennow K, Gatz M, et al. (1 January 2011). "Genetic association of sequence variants near AGER/NOTCH4 and dementia". Journal of Alzheimer's Disease. 24 (3): 475–84. doi:10.3233/jad-2011-101848. PMC 3477600. PMID 21297263.
- ↑ Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (January 1999). "KEGG: Kyoto Encyclopedia of Genes and Genomes". Nucleic Acids Research. 27 (1): 29–34. doi:10.1093/nar/27.1.29. PMC 148090. PMID 9847135.
- ↑ Vastrik I, D'Eustachio P, Schmidt E, Joshi-Tope G, Gopinath G, Croft D, et al. (2007). "Reactome: a knowledge base of biologic pathways and processes". Genome Biology. 8 (3): R39. doi:10.1186/gb-2007-8-3-r39. PMC 1868929. PMID 17367534.
- ↑ Pathway Studio Pathways
- ↑ Pathway Central
- ↑ Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. (2004). "Bioconductor: open software development for computational biology and bioinformatics". Genome Biology. 5 (10): R80. doi:10.1186/gb-2004-5-10-r80. PMC 545600. PMID 15461798.
- ↑ Dabbish, L., Stuart, C., Tsay, J., and Herbsleb, J. (2012). "Social coding in github: transparency and collaboration in an open software repository," in Proceedings of the ACM 2012 Conference on Computer Supported Cooperative Work (New York, NY: ACM), 1277–1286
- 1 2 3 Khatri P, Sirota M, Butte AJ (23 February 2012). "Ten years of pathway analysis: current approaches and outstanding challenges". PLOS Computational Biology. 8 (2): e1002375. Bibcode:2012PLSCB...8E2375K. doi:10.1371/journal.pcbi.1002375. PMC 3285573. PMID 22383865.
- 1 2 Henderson-Maclennan NK, Papp JC, Talbot CC, McCabe ER, Presson AP (2010). "Pathway analysis software: annotation errors and solutions". Molecular Genetics and Metabolism. 101 (2–3): 134–40. doi:10.1016/j.ymgme.2010.06.005. PMC 2950253. PMID 20663702.
- ↑ Emmert-Streib F, Dehmer M (May 2011). "Networks for systems biology: conceptual connection of data and function". IET Systems Biology. 5 (3): 185–207. doi:10.1049/iet-syb.2010.0025. PMID 21639592.
- 1 2 Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, et al. (October 2007). "A systems biology approach for pathway level analysis". Genome Research. 17 (10): 1537–45. doi:10.1101/gr.6202607. PMC 1987343. PMID 17785539.
- 1 2 Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, et al. (January 2009). "A novel signaling pathway impact analysis". Bioinformatics. 25 (1): 75–82. doi:10.1093/bioinformatics/btn577. PMC 2732297. PMID 18990722.
- ↑ Glaab E, Baudot A, Krasnogor N, Schneider R, Valencia A (September 2012). "EnrichNet: network-based gene set enrichment analysis". Bioinformatics. 28 (18): i451–i457. doi:10.1093/bioinformatics/bts389. PMC 3436816. PMID 22962466.
- ↑ Geistlinger L, Csaba G, Küffner R, Mulder N, Zimmer R (July 2011). "From sets to graphs: towards a realistic enrichment analysis of transcriptomic systems". Bioinformatics. 27 (13): i366-73. doi:10.1093/bioinformatics/btr228. PMC 3117393. PMID 21685094.
- ↑ Glaab E, Baudot A, Krasnogor N, Valencia A (May 2010). "TopoGSA: network topological gene set analysis". Bioinformatics. 26 (9): 1271–2. doi:10.1093/bioinformatics/btq131. PMC 2859135. PMID 20335277.
- ↑ Shojaie A, Michailidis G (22 May 2010). "Network enrichment analysis in complex experiments". Statistical Applications in Genetics and Molecular Biology. 9 (1): Article22. doi:10.2202/1544-6115.1483. PMC 2898649. PMID 20597848.
- ↑ Huttenhower C, Haley EM, Hibbs MA, Dumeaux V, Barrett DR, Coller HA, Troyanskaya OG (June 2009). "Exploring the human genome with functional maps". Genome Research. 19 (6): 1093–106. doi:10.1101/gr.082214.108. PMC 2694471. PMID 19246570.
- ↑ Alexeyenko A, Lee W, Pernemalm M, Guegan J, Dessen P, Lazar V, et al. (September 2012). "Network enrichment analysis: extension of gene-set enrichment analysis to gene networks". BMC Bioinformatics. 13: 226. doi:10.1186/1471-2105-13-226. PMC 3505158. PMID 22966941.
- ↑ Signorelli M, Vinciotti V, Wit EC (September 2016). "NEAT: an efficient network enrichment analysis test". BMC Bioinformatics. 17 (1): 352. arXiv:1604.01210. doi:10.1186/s12859-016-1203-6. PMC 5011912. PMID 27597310. S2CID 2274758.
- ↑ Jeggari A, Alexeyenko A (March 2017). "NEArender: an R package for functional interpretation of 'omics' data via network enrichment analysis". BMC Bioinformatics. 18 (Suppl 5): 118. doi:10.1186/s12859-017-1534-y. PMC 5374688. PMID 28361684.
- ↑ Hong MG, Alexeyenko A, Lambert JC, Amouyel P, Prince JA (October 2010). "Genome-wide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease". Journal of Human Genetics. 55 (10): 707–9. doi:10.1038/jhg.2010.92. PMID 20668461. S2CID 27020289.
- ↑ Jeggari A, Alekseenko Z, Petrov I, Dias JM, Ericson J, Alexeyenko A (July 2018). "EviNet: a web platform for network enrichment analysis with flexible definition of gene sets". Nucleic Acids Research. 46 (W1): W163–W170. doi:10.1093/nar/gky485. PMC 6030852. PMID 29893885.
- ↑ "Ingenuity IPA - Integrate and Understand Complex 'omics Data". Ingenuity. 8 April 2015.
- ↑ Pathway Studio
- ↑ Pathway Studio Viewer
- 1 2 Mitrea C, Taghavi Z, Bokanizad B, Hanoudi S, Tagett R, Donato M, et al. (October 2013). "Methods and approaches in the topology-based analysis of biological pathways". Frontiers in Physiology. 4: 278. doi:10.3389/fphys.2013.00278. PMC 3794382. PMID 24133454.
- ↑ Franco M, Jeggari A, Peuget S, Böttger F, Selivanova G, Alexeyenko A (February 2019). "Prediction of response to anti-cancer drugs becomes robust via network integration of molecular data". Scientific Reports. 9 (1): 2379. Bibcode:2019NatSR...9.2379F. doi:10.1038/s41598-019-39019-2. PMC 6382934. PMID 30787419.