
The perturbed γ-γ angular correlation, PAC for short or PAC-Spectroscopy, is a method of nuclear solid-state physics with which magnetic and electric fields in crystal structures can be measured. In doing so, electrical field gradients and the Larmor frequency in magnetic fields as well as dynamic effects are determined. With this very sensitive method, which requires only about 10–1000 billion atoms of a radioactive isotope per measurement, material properties in the local structure, phase transitions, magnetism and diffusion can be investigated. The PAC method is related to nuclear magnetic resonance and the Mössbauer effect, but shows no signal attenuation at very high temperatures. Today only the time-differential perturbed angular correlation (TDPAC) is used.
History and development

PAC goes back to a theoretical work by Donald R. Hamilton[1] from 1940. The first successful experiment was carried out by Brady and Deutsch[2] in 1947. Essentially spin and parity of nuclear spins were investigated in these first PAC experiments. However, it was recognized early on that electric and magnetic fields interact with the nuclear moment,[3] providing the basis for a new form of material investigation: nuclear solid-state spectroscopy.
Step by step the theory was developed.[4][5][6][7][8][9][10][11][12][13][14][15][16][17] After Abragam and Pound[18] published their work on the theory of PAC in 1953 including extra nuclear fields, many studies with PAC were carried out afterwards. In the 1960s and 1970s, interest in PAC experiments sharply increased, focusing mainly on magnetic and electric fields in crystals into which the probe nuclei were introduced. In the mid-1960s, ion implantation was discovered, providing new opportunities for sample preparation. The rapid electronic development of the 1970s brought significant improvements in signal processing. From the 1980s to the present, PAC has emerged as an important method for the study and characterization of materials,[19][20][21][22][23] e.g. for the study of semiconductor materials, intermetallic compounds, surfaces and interfaces, and a number of applications have also appeared in biochemistry.[24]
While until about 2008 PAC instruments used conventional high-frequency electronics of the 1970s, in 2008 Christian Herden and Jens Röder et al. developed the first fully digitized PAC instrument that enables extensive data analysis and parallel use of multiple probes.[25] Replicas and further developments followed.[26][27]
Measuring principle

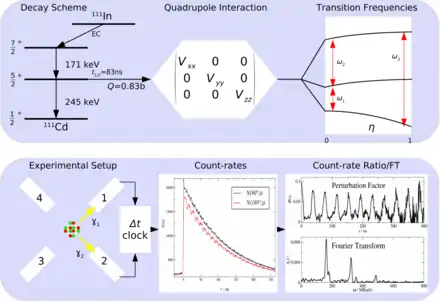
PAC uses radioactive probes, which have an intermediate state with decay times of 2 ns to approx. 10 μs, see example 111In in the picture on the right. After electron capture (EC), indium transmutates to cadmium. Immediately thereafter, the 111cadmium nucleus is predominantly in the excited 7/2+ nuclear spin and only to a very small extent in the 11/2- nuclear spin, the latter should not be considered further. The 7/2+ excited state transitions to the 5/2+ intermediate state by emitting a 171 keV γ-quantum. The intermediate state has a lifetime of 84.5 ns and is the sensitive state for the PAC. This state in turn decays into the 1/2+ ground state by emitting a γ-quantum with 245 keV. PAC now detects both γ-quanta and evaluates the first as a start signal, the second as a stop signal.

Now one measures the time between start and stop for each event. This is called coincidence when a start and stop pair has been found. Since the intermediate state decays according to the laws of radioactive decay, one obtains an exponential curve with the lifetime of this intermediate state after plotting the frequency over time. Due to the non-spherically symmetric radiation of the second γ-quantum, the so-called anisotropy, which is an intrinsic property of the nucleus in this transition, it comes with the surrounding electrical and/or magnetic fields to a periodic disorder (hyperfine interaction). The illustration of the individual spectra on the right shows the effect of this disturbance as a wave pattern on the exponential decay of two detectors, one pair at 90° and one at 180° to each other. The waveforms to both detector pairs are shifted from each other. Very simply, one can imagine a fixed observer looking at a lighthouse whose light intensity periodically becomes lighter and darker. Correspondingly, a detector arrangement, usually four detectors in a planar 90 ° arrangement or six detectors in an octahedral arrangement, "sees" the rotation of the core on the order of magnitude of MHz to GHz.

According to the number n of detectors, the number of individual spectra (z) results after z=n²-n, for n=4 therefore 12 and for n=6 thus 30. In order to obtain a PAC spectrum, the 90° and 180° single spectra are calculated in such a way that the exponential functions cancel each other out and, in addition, the different detector properties shorten themselves. The pure perturbation function remains, as shown in the example of a complex PAC spectrum. Its Fourier transform gives the transition frequencies as peaks.
, the count rate ratio, is obtained from the single spectra by using:


Depending on the spin of the intermediate state, a different number of transition frequencies show up. For 5/2 spin, 3 transition frequencies can be observed with the ratio ω1+ω2=ω3. As a rule, a different combination of 3 frequencies can be observed for each associated site in the unit cell.

PAC is a statistical method: Each radioactive probe atom sits in its own environment. In crystals, due to the high regularity of the arrangement of the atoms or ions, the environments are identical or very similar, so that probes on identical lattice sites experience the same hyperfine field or magnetic field, which then becomes measurable in a PAC spectrum. On the other hand, for probes in very different environments, such as in amorphous materials, a broad frequency distribution or no is usually observed and the PAC spectrum appears flat, without frequency response. With single crystals, depending on the orientation of the crystal to the detectors, certain transition frequencies can be reduced or extinct, as can be seen in the example of the PAC spectrum of zinc oxide (ZnO).
Instrumental setup
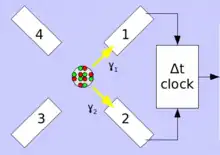

In the typical PAC spectrometer, a setup of four 90° and 180° planar arrayed detectors or six octahedral arrayed detectors are placed around the radioactive source sample. The detectors used are scintillation crystals of BaF2 or NaI. For modern instruments today mainly LaBr3:Ce or CeBr3 are used. Photomultipliers convert the weak flashes of light into electrical signals generated in the scintillator by gamma radiation. In classical instruments these signals are amplified and processed in logical AND/OR circuits in combination with time windows the different detector combinations (for 4 detectors: 12, 13, 14, 21, 23, 24, 31, 32, 34, 41, 42, 43) assigned and counted. Modern digital spectrometers use digitizer cards that directly use the signal and convert it into energy and time values and store them on hard drives. These are then searched by software for coincidences. Whereas in classical instruments, "windows" limiting the respective γ-energies must be set before processing, this is not necessary for the digital PAC during the recording of the measurement. The analysis only takes place in the second step. In the case of probes with complex cascades, this makes it makes it possible to perform a data optimization or to evaluate several cascades in parallel, as well as measuríng different probes simultaneously. The resulting data volumes can be between 60 and 300 GB per measurement.
Sample materials
As materials for the investigation (samples) are in principle all materials that can be solid and liquid. Depending on the question and the purpose of the investigation, certain framework conditions arise. For the observation of clear perturbation frequencies it is necessary, due to the statistical method, that a certain proportion of the probe atoms are in a similar environment and e.g. experiences the same electric field gradient. Furthermore, during the time window between the start and stop, or approximately 5 half-lives of the intermediate state, the direction of the electric field gradient must not change. In liquids, therefore, no interference frequency can be measured as a result of the frequent collisions, unless the probe is complexed in large molecules, such as in proteins. The samples with proteins or peptides are usually frozen to improve the measurement.
The most studied materials with PAC are solids such as semiconductors, metals, insulators, and various types of functional materials. For the investigations, these are usually crystalline. Amorphous materials do not have highly ordered structures. However, they have close proximity, which can be seen in PAC spectroscopy as a broad distribution of frequencies. Nano-materials have a crystalline core and a shell that has a rather amorphous structure. This is called core-shell model. The smaller the nanoparticle becomes, the larger the volume fraction of this amorphous portion becomes. In PAC measurements, this is shown by the decrease of the crystalline frequency component in a reduction of the amplitude (attenuation).
Sample preparation
The amount of suitable PAC isotopes required for a measurement is between about 10 to 1000 billion atoms (1010-1012). The right amount depends on the particular properties of the isotope. 10 billion atoms are a very small amount of substance. For comparison, one mol contains about 6.22x1023 particles. 1012 atoms in one cubic centimeter of beryllium give a concentration of about 8 nmol/L (nanomol=10−9 mol). The radioactive samples each have an activity of 0.1-5 MBq, which is in the order of the exemption limit for the respective isotope.
How the PAC isotopes are brought into the sample to be examined is up to the experimenter and the technical possibilities. The following methods are usual:
Implantation

During implantation, a radioactive ion beam is generated, which is directed onto the sample material. Due to the kinetic energy of the ions (1-500 keV) these fly into the crystal lattice and are slowed down by impacts. They either come to a stop at interstitial sites or push a lattice-atom out of its place and replace it. This leads to a disruption of the crystal structure. These disorders can be investigated with PAC. By tempering these disturbances can be healed. If, on the other hand, radiation defects in the crystal and their healing are to be examined, unperseived samples are measured, which are then annealed step by step.
The implantation is usually the method of choice, because it can be used to produce very well-defined samples.
Evaporation
In a vacuum, the PAC probe can be evaporated onto the sample. The radioactive probe is applied to a hot plate or filament, where it is brought to the evaporation temperature and condensed on the opposite sample material. With this method, e.g. surfaces are examined. Furthermore, by vapor deposition of other materials, interfaces can be produced. They can be studied during tempering with PAC and their changes can be observed. Similarly, the PAC probe can be transferred to sputtering using a plasma.
Diffusion
In the diffusion method, the radioactive probe is usually diluted in a solvent applied to the sample, dried and it is diffused into the material by tempering it. The solution with the radioactive probe should be as pure as possible, since all other substances can diffuse into the sample and affect thereby the measurement results. The sample should be sufficiently diluted in the sample. Therefore, the diffusion process should be planned so that a uniform distribution or sufficient penetration depth is achieved.
Added during synthesis
PAC probes may also be added during the synthesis of sample materials to achieve the most uniform distribution in the sample. This method is particularly well suited if, for example, the PAC probe diffuses only poorly in the material and a higher concentration in grain boundaries is to be expected. Since only very small samples are necessary with PAC (about 5 mm), micro-reactors can be used. Ideally, the probe is added to the liquid phase of the sol-gel process or one of the later precursor phases.
Neutron activation
In neutron activation, the probe is prepared directly from the sample material by converting very small part of one of the elements of the sample material into the desired PAC probe or its parent isotope by neutron capture. As with implantation, radiation damage must be healed. This method is limited to sample materials containing elements from which neutron capture PAC probes can be made. Furthermore, samples can be intentionally contaminated with those elements that are to be activated. For example, hafnium is excellently suited for activation because of its large capture cross section for neutrons.
Nuclear reaction
Rarely used are direct nuclear reactions in which nuclei are converted into PAC probes by bombardment by high-energy elementary particles or protons. This causes major radiation damage, which must be healed. This method is used with PAD, which belongs to the PAC methods.
Laboratories
The currently largest PAC laboratory in the world is located at ISOLDE in CERN with about 10 PAC instruments, that receives its major funding form BMBF. Radioactive ion beams are produced at the ISOLDE by bombarding protons from the booster onto target materials (uranium carbide, liquid tin, etc.) and evaporating the spallation products at high temperatures (up to 2000 °C), then ionizing them and then accelerating them. With the subsequent mass separation usually very pure isotope beams can be produced, which can be implanted in PAC samples. Of particular interest to the PAC are short-lived isomeric probes such as: 111mCd, 199mHg, 204mPb, and various rare earth probes.
Theory


The first -quantum () will be emitted isotropically. Detecting this quantum in a detector selects a subset with an orientation of the many possible directions that has a given. The second -quantum () has an anisotropic emission and shows the effect of the angle correlation. The goal is to measure the relative probability with the detection of at the fixed angle in relation to . The probability is given with the angle correlation (perturbation theory):








For a --cascade, is due to the preservation of parity:


Where is the spin of the intermediate state and with the multipolarity of the two transitions. For pure multipole transitions, is .




is the anisotropy coefficient that depends on the angular momentum of the intermediate state and the multipolarities of the transition.

The radioactive nucleus is built into the sample material and emits two -quanta upon decay. During the lifetime of the intermediate state, i.e. the time between and , the core experiences a disturbance due to the hyperfine interaction through its electrical and magnetic environment. This disturbance changes the angular correlation to:

is the perturbation factor. Due to the electrical and magnetic interaction, the angular momentum of the intermediate state experiences a torque about its axis of symmetry. Quantum-mechanically, this means that the interaction leads to transitions between the M states. The second -quantum () is then sent from the intermediate level. This population change is the reason for the attenuation of the correlation.

The interaction occurs between the magnetic core dipole moment and the intermediate state or/and an external magnetic field . The interaction also takes place between nuclear quadrupole moment and the off-core electric field gradient .



Magnetic dipole interaction
For the magnetic dipole interaction, the frequency of the precession of the nuclear spin around the axis of the magnetic field is given by:


is the Landé g-factor und is the nuclear magneton.


With follows:


From the general theory we get:

For the magnetic interaction follows:

Static electric quadrupole interaction
The energy of the hyperfine electrical interaction between the charge distribution of the core and the extranuclear static electric field can be extended to multipoles. The monopole term only causes an energy shift and the dipole term disappears, so that the first relevant expansion term is the quadrupole term:
- ij=1;2;3

This can be written as a product of the quadrupole moment and the electric field gradient . Both [tensor]s are of second order. Higher orders have too small effect to be measured with PAC.


The electric field gradient is the second derivative of the electric potential at the core:


becomes diagonalized, that:

The matrix is free of traces in the main axis system (Laplace equation)

Typically, the electric field gradient is defined with the largest proportion and :

- ,


In cubic crystals, the axis parameters of the unit cell x, y, z are of the same length. Therefore:
- and


In axisymmetric systems is .
For axially symmetric electric field gradients, the energy of the substates has the values:

The energy difference between two substates, and , is given by:



The quadrupole frequency is introduced. The formulas in the colored frames are important for the evaluation:



The publications mostly list . as elementary charge and as Planck constant are well known or well defined. The nuclear quadrupole moment is often determined only very inaccurately (often only with 2-3 digits). Because can be determined much more accurately than , it is not useful to specify only because of the error propagation. In addition, is independent of spin! This means that measurements of two different isotopes of the same element can be compared, such as 199mHg(5/2−), 197mHg(5/2−) and 201mHg(9/2−). Further, can be used as finger print method.




For the energy difference then follows:

If , then:

with:

For integer spins applies:
- und


For half integer spins applies:
- und


The perturbation factor is given by:

With the factor for the probabilities of the observed frequencies:

As far as the magnetic dipole interaction is concerned, the electrical quadrupole interaction also induces a precision of the angular correlation in time and this modulates the quadrupole interaction frequency. This frequency is an overlap of the different transition frequencies . The relative amplitudes of the various components depend on the orientation of the electric field gradient relative to the detectors (symmetry axis) and the asymmetry parameter . For a probe with different probe nuclei, one needs a parameter that allows a direct comparison: Therefore, the quadrupole coupling constant independent of the nuclear spin is introduced.


Combined interactions
If there is a magnetic and electrical interaction at the same time on the radioactive nucleus as described above, combined interactions result. This leads to the splitting of the respectively observed frequencies. The analysis may not be trivial due to the higher number of frequencies that must be allocated. These then depend in each case on the direction of the electric and magnetic field to each other in the crystal. PAC is one of the few ways in which these directions can be determined.
Dynamic interactions
If the hyperfine field fluctuates during the lifetime of the intermediate level due to jumps of the probe into another lattice position or from jumps of a near atom into another lattice position, the correlation is lost. For the simple case with an undistorted lattice of cubic symmetry, for a jump rate of for equivalent places , an exponential damping of the static -terms is observed:






Here is a constant to be determined, which should not be confused with the decay constant . For large values of , only pure exponential decay can be observed:




The boundary case after Abragam-Pound is , if , then:


After effects

Cores that transmute beforehand of the --cascade usually cause a charge change in ionic crystals (In3+) to Cd2+). As a result, the lattice must respond to these changes. Defects or neighboring ions can also migrate. Likewise, the high-energy transition process may cause the Auger effect, that can bring the core into higher ionization states. The normalization of the state of charge then depends on the conductivity of the material. In metals, the process takes place very quickly. This takes considerably longer in semiconductors and insulators. In all these processes, the hyperfine field changes. If this change falls within the --cascade, it may be observed as an after effect.
The number of nuclei in state (a) in the image on the right is depopulated both by the decay after state (b) and after state (c):

mit:

From this one obtains the exponential case:

For the total number of nuclei in the static state (c) follows:

The initial occupation probabilities are for static and dynamic environments:



General theory
In the general theory for a transition is given:




- Minimum von







with:

References
- ↑ Hamilton, Donald R. (1940-07-15). "On Directional Correlation of Successive Quanta". Physical Review. American Physical Society (APS). 58 (2): 122–131. Bibcode:1940PhRv...58..122H. doi:10.1103/physrev.58.122. ISSN 0031-899X.
- ↑ Brady, Edward L.; Deutsch, Martin (1947-11-01). "Angular Correlation of Successive Gamma-Ray Quanta". Physical Review. American Physical Society (APS). 72 (9): 870–871. Bibcode:1947PhRv...72..870B. doi:10.1103/physrev.72.870. ISSN 0031-899X.
- ↑ Aeppli, H.; Bishop, A. S.; Frauenfelder, H.; Walter, M.; Zünti, W. (1951-05-15). "Influence of the Atomic Shell on Nuclear Angular Correlation in Cd111". Physical Review. American Physical Society (APS). 82 (4): 550. Bibcode:1951PhRv...82..550A. doi:10.1103/physrev.82.550. ISSN 0031-899X.
- ↑ Gardner, J W (1949-12-01). "Directional Correlation between Successive Internal-Conversion Electrons". Proceedings of the Physical Society. Section A. IOP Publishing. 62 (12): 763–779. Bibcode:1949PPSA...62..763G. doi:10.1088/0370-1298/62/12/302. ISSN 0370-1298.
- ↑ Ling, Daniel S.; Falkoff, David L. (1949-12-01). "Interference Effects in Gamma-Gamma Angular Correlations". Physical Review. American Physical Society (APS). 76 (11): 1639–1648. Bibcode:1949PhRv...76.1639L. doi:10.1103/physrev.76.1639. ISSN 0031-899X.
- ↑ Fierz, M. (1949). "Zur Theorie der Multipolstrahlung". Helvetica Physica Acta (in German). 22 (4): 489.
- ↑ J.A. Spiers, Nat. Res. Council Canada, Publ. No. 1925 (1950)
- ↑ Spiers, J. A. (1950-11-01). "On the Directional Correlation of Successive Nuclear Radiations". Physical Review. American Physical Society (APS). 80 (3): 491. Bibcode:1950PhRv...80..491S. doi:10.1103/physrev.80.491. ISSN 0031-899X.
- ↑ Falkoff, David L.; Uhlenbeck, G. E. (1950-07-15). "On the Directional Correlation of Successive Nuclear Radiations". Physical Review. American Physical Society (APS). 79 (2): 323–333. Bibcode:1950PhRv...79..323F. doi:10.1103/physrev.79.323. ISSN 0031-899X.
- ↑ Racah, Giulio (1951-12-01). "Directional Correlation of Successive Nuclear Radiations". Physical Review. American Physical Society (APS). 84 (5): 910–912. Bibcode:1951PhRv...84..910R. doi:10.1103/physrev.84.910. ISSN 0031-899X.
- ↑ U. Fano, Nat'l. Bureau of Standards Report 1214
- ↑ Fano, U. (1953-05-15). "Geometrical Characterization of Nuclear States and the Theory of Angular Correlations". Physical Review. American Physical Society (APS). 90 (4): 577–579. Bibcode:1953PhRv...90..577F. doi:10.1103/physrev.90.577. ISSN 0031-899X.
- ↑ Lloyd, Stuart P. (1952-03-01). "The Angular Correlation of Two Successive Nuclear Radiations". Physical Review. American Physical Society (APS). 85 (5): 904–911. Bibcode:1952PhRv...85..904L. doi:10.1103/physrev.85.904. ISSN 0031-899X.
- ↑ Adler, K. (1952). "Beiträge zur Theorie der Richtungskorrelation". Helvetica Physica Acta (in German). 25 (3): 235.
- ↑ De Groot, S.R. (1952). "On the theories of angular distribution and correlation of beta and gamma radiation". Physica. Elsevier BV. 18 (12): 1201–1214. Bibcode:1952Phy....18.1201D. doi:10.1016/s0031-8914(52)80196-x. ISSN 0031-8914.
- ↑ F. Coester, J.M. Jauch, Helv. Phys. Acta 26 (1953) 3.
- ↑ Biedenharn, L. C.; Rose, M. E. (1953-07-01). "Theory of Angular Correlation of Nuclear Radiations". Reviews of Modern Physics. American Physical Society (APS). 25 (3): 729–777. Bibcode:1953RvMP...25..729B. doi:10.1103/revmodphys.25.729. ISSN 0034-6861.
- ↑ Abragam, A.; Pound, R. V. (1953-11-15). "Influence of Electric and Magnetic Fields on Angular Correlations". Physical Review. American Physical Society (APS). 92 (4): 943–962. Bibcode:1953PhRv...92..943A. doi:10.1103/physrev.92.943. ISSN 0031-899X.
- ↑ Th. Wichert, E. Recknagel: Perturbed Angular Correlation. In: Ulrich Gonser (Hrsg.): Microscopic Methods in Metals (= Topics in Current Physics. Band 40). Springer, Berlin/Heidelberg 1986, ISBN 978-3-642-46571-0, S. 317–364, doi:10.1007/978-3-642-46571-0_11
- ↑ Collins, Gary S.; Shropshire, Steven L.; Fan, Jiawen (1990). "Perturbed γ−γ angular correlations: A spectroscopy for point defects in metals and alloys". Hyperfine Interactions. Springer Science and Business Media LLC. 62 (1–2): 1–34. doi:10.1007/bf02407659. ISSN 0304-3843. S2CID 94593348.
- ↑ Th. Wichert, N. Achziger, H. Metzner, R. Sielemann: Perturbed angular correlation. In: G. Langouche (Hrsg.): Hyperfine Interactions of Defects in Semiconductors. Elsevier, Amsterdam 1992, ISBN 0-444-89134-X, S. 77
- ↑ Jens Röder, Klaus-dieter Becker: Perturbed γ–γ Angular Correlation. In: Methods in Physical Chemistry. John Wiley & Sons, Ltd, 2012, ISBN 978-3-527-32745-4, S. 325–349, doi:10.1002/9783527636839.ch10
- ↑ Günter Schatz, Alois Weidinger, Manfred Deicher: Nukleare Festkörperphysik: Kernphysikalische Messmethoden und ihre Anwendungen. 4. Auflage. Vieweg+Teubner Verlag, 2010, ISBN 978-3-8351-0228-6
- ↑ Hemmingsen, Lars; Sas, Klára Nárcisz; Danielsen, Eva (2004). "Biological Applications of Perturbed Angular Correlations of γ-Ray Spectroscopy". Chemical Reviews. American Chemical Society (ACS). 104 (9): 4027–4062. doi:10.1021/cr030030v. ISSN 0009-2665. PMID 15352785.
- ↑ Herden, C.; Röder, J.; Gardner, J.A.; Becker, K.D. (2008). "Fully digital time differential perturbed angular correlation (TDPAC) spectrometer". Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. Elsevier BV. 594 (2): 155–161. Bibcode:2008NIMPA.594..155H. doi:10.1016/j.nima.2008.05.001. ISSN 0168-9002.
- ↑ Nagl, Matthias; Vetter, Ulrich; Uhrmacher, Michael; Hofsäss, Hans (2010). "A new all-digital time differential γ-γ angular correlation spectrometer". Review of Scientific Instruments. AIP Publishing. 81 (7): 073501–073501–9. Bibcode:2010RScI...81g3501N. doi:10.1063/1.3455186. ISSN 0034-6748. PMID 20687716.
- ↑ Jäger, M.; Iwig, K.; Butz, T. (2010). "A user-friendly fully digital TDPAC-spectrometer". Hyperfine Interactions. Springer Science and Business Media LLC. 198 (1–3): 167–172. Bibcode:2010HyInt.198..167J. doi:10.1007/s10751-010-0201-8. ISSN 0304-3843. S2CID 17531166.