
Phosphasilenes or silylidenephosphanes are a class of compounds with silicon-phosphorus double bonds.[1] Since the electronegativity of phosphorus (2.1) is higher than that of silicon (1.9), the "Si=P" moiety of phosphasilene is polarized.[2][3] The degree of polarization can be tuned by altering the coordination numbers of the Si and P centers, or by modifying the electronic properties of the substituents.[2] The phosphasilene Si=P double bond is highly reactive, yet with the choice of proper substituents, it can be stabilized via donor-acceptor interaction or by steric congestion.[3]

The landmark discovery of the first phosphasilene by NMR spectroscopy was made in 1984 by Bickelhaupt et al.[4] The first phosphasilene came with bulky aryl substituents at the phosphorus and silicon atoms.[4] Almost a decade after this spectroscopic observation, the first structural characterization of phosphasilene was achieved in 1993 by Niecke et al.[5] The successful isolation of phosphasilenes with silicon-phosphorus double bonds represents one of the discoveries that challenged and disproved the "double-bond rule".
Synthesis
Synthesis via β-elimination

An important synthetic pathway towards phosphasilene is the 1,2-elimination reactions of silylphosphane derivatives. The first metastable arylphosphasilenes were accessed by Bickelhaupt et al. via the deprotonation of in situ formed (chlorosilyl)phosphanes ArP(H)–(Cl)SiAr2 using organolithium bases.[2][4][6][7] As shown in the scheme on the left, it is also possible to use pre-formed lithium phosphanides Ar'P(H)Li as both a phosphorus source and a base. However, the latter synthetic pathway involves formation of primary phosphines ArPH2, which can be difficult of be separated from phosphasilenes.[2] Despite such a drawback, this strategy has been successfully applied by Niecke et al. to obtain a series of 1,3-diphospha-2-silaallyl anions, which serve as precursors for 2-phosphanylphosphasilenes.[2][8]

Applying an analogous strategy, Driess and coworkers developed an effective approach for synthesizing P-silyl phosphasilenes via the thermal elimination of LiF from corresponding lithium (flouorosilyl)phosphanides.[9][10][11]
Synthesis based on reactivities of stable silyene

Phosphasilenes with 4-coordinate silicon, which can also be viewed as silylene-stabilized phosphinidene, can be synthesized based on the reactivities of stable silyene complexes.[2] For example, Inoue and coworkers demonstrated that benzamidinate-stabilized phosphanylsilylene can give rise to corresponding Si- and P-trimethylsilyl-substituted phosphasilene via thermal rearrangement, while the reaction can also yield 4-disila-1,3-diphosphacyclobuta-diene with the addition of a mild chlorinating agent Ph3PCl2.[12][2] In these phosphasilenes with four-coordinate silicon, even though formally five bonds are drawn around silicon, the Si–P π bonds are calculated to be strongly polarized towards the P atoms. The contribution from phosphorus and silicon to the π orbitals are calculated to be 87.53% and 12.47%, respectively.
Structure and bonding
The parent phosphasilene H2Si=PH

The unstable parent phosphasilene H2Si=PH has been generated in the gas phase by the reacting atomic silicon with phosphine PH3, and identified via matrix isolation spectroscopy methods.[14] Density functional theory (DFT) calculations suggests that in the ground state, H2Si=PH exists in a singlet spin state, with Cs-symmetric planar geometry.[14] At the B3LYP/6-311+G** level of theory, the Si=P bond length and the Si-P-H bond angle are calculated to be 2.084 Å and 90.7o.[14] The Si=P bond dissociation energy is 75.0 kcal mol−1 at the B97-D/6-31G(d) level of theory; while the π-bond energy, Dπ(Si=P) is 36.6/35.9 kcal mol−1.[13] The frontier orbitals of the parent phosphasilene consists of the π bonding and π anti-bonding orbitals: π(Si=P) and π*(Si=P) correspond to the HOMO and LUMO, respectively. HOMO-1 was calculated to be the lone pair on phosphorus n(P).[15]
"Half-Parent" Phosphasilene R2Si=PH
Driess and coworkers prepared thermally stable "half"-parent phosphasilene R2Si=PH (R2Si = (tBu3Si)(iPr3C6H2)Si), which is the first example of phosphasilene with a terminal PH group.[16] This species was obtained as a mixture of E/Z isomers, thus its 31P NMR spectrum featured two doublets with 29Si satellites (δ=123.1, 1J (P, H)=123 Hz, 1J (P, Si)=157 Hz and δ=134.2 ppm, 1J (P, H)=131 Hz, 1J (P, Si)=130 Hz).[16] These 1J (P, H) coupling constants are much smaller compared to those of secondary phosphanes (R2PH) and phosphaalkenes with a PH group, which indicates that the phosphasilene phosphorus atom possesses more 3p character.[16] X-ray crystallography of this "half"-parent phosphasilene species shows that the silicon atom occupies a trigonal-planar coordination environment.[16] The Si=P bond distance was reported to be 2.094(5) Å, which is about 7% shorter than a typical silicon-phosphorus single bond, but only slightly longer than that of P-silyl-substituted phosphasilenes,[16] which suggests that the potential of Si=P bond on a potential energy surface is relatively shallow.[16][17]
π-conjugated phosphasilenes

Tamao et al. reported a series of π-conjugated phosphasilenes stabilized by Eind groups (Eind=1,1,3,3,5,5,7,7-octaethyl-s-hydrindacen-4-yl).[18] These systems feature Si=P units that are highly coplanar with the aromatic ring, allowing strong π → π* absorptions. The coplanarity is made possible by the rigidity of the two Eind groups that are oriented trans and perpendicular with respect to the Si=P bond.[18] The Si=P bond length observed by X-ray crystallography are ca. 2.09-2.10 Å, which are typical for phosphasilenes.[18]

The bonding of π-conjugated phosphasilenes has been probed by DFT calculations at the B3LYP/6-31G** level. The HOMO was calculated to represent mostly the 3pπ(Si–P), while the LUMO featured significant contribution from the 3pπ*(Si–P)–2pπ*(phenyl) conjugation. The HOMO-1 orbital involves the 3n–2pπ conjugation, which originate from the presence of lone pair on the phosphorus atom and the π-orbital on the Eind benzene ring.[18]
"Push-pull" phosphasilene
By installing electron-donating substituents on silicon and electron-withdrawing substituents on phosphorus, the Si=P bond polarization can be decreased and even reversed through the "push-pull" interaction of the substituents with opposing electronic effects.[19] Applying this design strategy, Escudié et al. prepared stable "push-pull" phosphasilene (tBu2MeSi)2Si=PMes* (Mes* = 2,4,6-tri-tertbutylphenyl) with electron-donating silyl groups on Si and an electron-withdrawing aryl group on P.[19] Computations on the model compound (Me3Si)2Si=PMes (Mes = 2,4,6-trimethylphenyl) demonstrate that n(P) and π*(Si=P) correspond to the HOMO and LUMO, respectively.[19] The relatively small energy gap between the interacting occupied (n(P)) and vacant (π*(Si=P)) molecular orbitals gives rise to a large paramagnetic contribution,[20] which explains the extreme deshielding of the doubly bonded Si and P atoms, as well as the red shift in the UV spectrum that are observed in (tBu2MeSi)2Si=PMes*.[19] In the "push-pull" phosphasilene prepared by Escudié and co-workers, the Si=P bond length was reported to be 2.1114(7) Å, which is longer than what was observed in most other reported phosphasilenes (2.062(1)–2.094(5) Å).[19]
Metallophosphasilenes R2Si=PM
Driess et al. demonstrated that stable metallophosphasilenes of the type R2Si=PM can be prepared from metalation reaction of "half"-parent phosphasilenes R2Si=PH (R2Si = (tBu3Si)(iPr3C6H2)Si).[16] NMR spectroscopic studies demonstrated that metalation led to the deshielding of the 31P nucleus, while the low coordinate 29Si atom in the metallophosphasilene became more shielded. Driess and coworkers explained this observation by proposing that the stabilization of the non-bonding orbital at phosphorus through n(P) → σ*(Si-Si) hyperconjugation is more effective after metalation.[16] This is due to the higher negative partial charge at the phosphorus atom in the metallophosphasilene. As shown in the scheme below, this shielding effects is analogous to what has been observed for related alkali-metal-substituted disilenylides of the type [M(R3Si)Si=Si(SiR3)2].[16][21][22] The Si=P distance in the metallophosphasilene that Driess et al. synthesized was reported to be 2.064(1) Å, which is significantly shorter than that of the "half"-parent compound (R2Si=PH) from which R2Si=PM was derived.[16] This contraction of the Si=P bond, together with a slight elongation of the Si–Si bond and a shrinkage of the P-Si-Si angle, has been rationalized by the increased hyperconjugative interactions in the R2Si=PM system.[16]
Phosphasilene with amino-substituents at Si
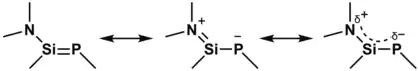
Due to the n(N)–π*(Si=P) orbital interaction, there exists strong delocalization of electron density from nitrogen to phosphorus in phosphasilene with amino-substituents at silicon.[2] Among the resonance structures shown in the figure on the left, the structure in the middle with zwitterionic structure was calculated to have a significant contribution.[2] Therefore, compared to the parent phosphasilene, the double-bond character of the Si–P is significantly reduced in phosphasilene with amino-substituents at Si, giving rise to longer Si–P bond distance.
Reactivity
Reaction with Lewis acids and bases
Due to the higher electronegativity of phosphorus and the polarized nature of the Si=P moiety, phosphasilene tend to react with Lewis acids and bases at the phosphorus atom and silicon atom, respectively. The reaction of phosphasilene with Lewis acids, which usually occurs at the Lewis-basic two-coordinate phosphorus atom, is also dependent upon the nature of the reactants.[2]
Lewis bases have been shown to react with both three-coordinate and four-coordinate silicon atoms. The coordination of Lewis bases to the three-coordinate silicon atom of phosphasilene can lead to effective stabilization of the Si=P moiety.[2] For example, it has been demonstrated that unstable phosphasilene can react with DMAP and small N-heterocyclic carbene (NHC) to afford the corresponding stable complexes.[23] In phosphasilenes stabilized by NHC, the Si=P bonds are elongated and the negative charge gets localized on the phosphorus atom. Among the resonance structures shown in the figure below, Natural Resonance Theory (NRT) indicates that the third structure with a Si–P single bond is predominant, carrying a resonance weight of 76.3%.[2][24] Therefore, these stabilized phosphasilene can also be interpreted as a silyene-phosphinidene adduct.[2]

For donor-stabilized phosphasilene with four coordinate silicon, the reaction with Lewis bases may lead to ligand exchange at the silicon atom.[25]
Metalation
Metalation of phosphasilenes gives rise to either complexes featuring the coordination of the phosphorus lone pair to a metal center [26][3][24] or P-metalated phosphasilenes.[27][16][28] In the former case, the binding of the phosphasilenes to transition metals via the phosphorus lone pair reduces the double-bond character of the Si–P bond.[2] Some examples of this type of phosphasilene transition metal complexes are shown below.
Driess and coworkers first observed the formation of P-metalated phosphasilenes of the latter case: P-ferrio-substituted phosphasilene R2Si=P[Fe(CO)2(η5-C5H5)] (R = 2,4,6-iPr3C6H2).[27] They further demonstrated that P-metalated phosphasilenes R2Si=PM can be obtained by metalating "half"-parent phosphasilenes, which substitutes the R2Si=PH hydrogen atom with transition metal-containing fragments.[3][16]
Phosphinidene transfer
Phosphasilenes with significant zwitterionic characters undergoes facile hemolytic cleavage of the fragile Si=P bond. This can be utilized for the liberation and transfer of phosphinidene (:PH) to unsaturated organic molecules.[29] Driess et al. demonstrated that a fragile "half"-parent phosphasilene LSi=PH (L = CH[(C=CH2)CMe(NAr)2]; Ar = 2,6-iPr2C6H3) with highly shielded PH moiety is capable of transferring :PH to NHC.[29]

Theoretical investigation by DFT (B3LYP/6-31G(d) level) revealed that this phosphasilene bears two highly localized lone pairs on the phosphorus atom due to the LSi=PH ↔ LSi–P+H− resonance. Based on natural bond orbital (NBO) analysis, the σ bond of Si=P involves even contributions from Si and P, while the π bond (HOMO-1) is strongly polarized to the phosphorus atom. This indicates that the π bond between silicon and phosphorus is not predominant, supporting the significance of the zwitterionic resonance structure in the description of Si–P bonding.[29]
Small molecule activation

The Si=P moiety of phosphasilene has been reported to demonstrate small molecule activation reactivities analogous to those observed in Si=Si, P=C, and other heavier alkene analogues. For example, phosphasilene with silyl substituents has been shown to activate white phosphorus (P4) under relatively mild reaction conditions to form 1,2,3-triphospha-4-silabicyclo[1.1.0]butanes,[9] which is similar to disilenes' reactivity.[31][2] Analogous to the behavior of phosphaalkenes,[32] phosphasilene can also activate chalcogens such as S8 and Te to form unstable three-membered ring compounds.[7]

Using a fragile zwitterionic "half-parent" phosphasilene L'Si=PH (L' = CH[(C=CH2)CMe(NAr)2], Ar = 2,6-iPr2C6H3), Driess and coworkers demonstrated that an unusual N-H activation reactivity can be achieved by the Si=P moiety in ammonia, affording L'Si(NH2)PH2 species.[23] This represent a rare example of catalyst-free 1,2-hydroamination reaction that's been reported in heavier alkene analogues.[2][33][34]
References
- ↑ Driess, M. (1994). "Some Aspects of the Chemistry of Silylidene-Phosphanes and -Arsanes". Coord. Chem. Rev. 145: 1–25. doi:10.1016/0010-8545(95)90212-0.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Nesterov, V.; Breit, N.; Inoue, S. (2017). "Advances in Phosphasilene Chemistry". Chem. Eur. J. 23 (50): 12014–12039. doi:10.1002/chem.201700829. PMID 28379639.
- 1 2 3 4 5 Hansen, K.; Szilvási, T.; Blom, B.; Driess, M. (2015). "Transition Metal Complexes of a "Half-Parent" Phosphasilene Adduct Representing Silylene→Phosphinidene→Metal Complexes". Organometallics. 34 (24): 5703–5708. doi:10.1021/acs.organomet.5b00772.
- 1 2 3 4 5 Smit, C. N.; Lock, F. M.; Bickelhaupt, F. (1984). "2,4,6-Tri-tert-butylphenylphosphene-Dimesitylsilene; The First Phosphasilaalkene". Tetrahedron Lett. 25 (28): 3011–3014. doi:10.1016/S0040-4039(01)81351-4.
- ↑ Bender, H. R. G.; Niecke, E.; Nieger, M. (1993). "The First X-ray Structure of a Phosphasilene: 1,3,4-Triphospha-2-silabutene-(1)". J. Am. Chem. Soc. 115 (8): 3314–3315. doi:10.1021/ja00061a034.
- 1 2 Smit, C. N.; Bickelhaupt, F. (1987). "Phosphasilenes: synthesis and spectroscopic characterization". Organometallics. 6 (6): 1156–1163. doi:10.1021/om00149a004.
- 1 2 3 van den Winkel, Y.; Bastiaans, H. M. M.; Bickelhaupt, F. (1991). "Phosphasilene synthesis and reactivity: an improved route to 1-(2,4,6-tri-tert-butylphenyl)-2-tert-butyl-2-(2,4,6-tri-isopropylphenyl)phosphasilene". J. Organomet. Chem. 405 (2): 183–194. doi:10.1016/0022-328X(91)86271-Q.
- 1 2 Lange, D.; Klein, E.; Bender, H.; Niecke, E.; Nieger, M. Pietschnig, R. (1998). "1,3-Diphospha-2-silaallylic Lithium Complexes and Anions: Synthesis, Crystal Structures, Reactivity, and Bonding Properties". Organometallics. 17 (12): 2425–2432. doi:10.1021/om9707937.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - 1 2 3 Driess, M. (1991). "1,2,3‐Triphospha‐4‐silabicyclo[l.1.0]butanes from Activated, Stable Phosphasilenes and White Phosphorus". Angew. Chem. Int. Ed. Engl. 30 (8): 1022–1024. doi:10.1002/anie.199110221.
- 1 2 Driess, M.; Pritzkow, H.; Rell, S.; Winkler, U. (1996). "Synthesis and Unusual Reactivity of Compounds Containing Silicon−Phosphorus and Silicon−Arsenic Double Bonds: New Silylidenephosphanes and -arsanes of the Type R2SiE(SiR3) (E = P, As)". Organometallics. 15 (7): 1845–1855. doi:10.1021/om9508851.
- ↑ Driess, M.; Rell, S.; Pritzkow, H. (1995). "First structural characterization of silicon–arsenic and silicon–phosphorus multiple bonds in silylated silylidene-arsanes and -phosphanes; X-ray structure of a tellura-arsasilirane derivative". J. Chem. Soc., Chem. Commun. 15 (2): 253–254. doi:10.1039/C39950000253.
- 1 2 Inoue, S.; Wang, W.; Präsang, C.; Asay, M.; Irran, E.; Driess, M. (2011). "An Ylide-like Phosphasilene and Striking Formation of a 4π-Electron, Resonance-Stabilized 2,4-Disila-1,3-diphosphacyclobutadiene". J. Am. Chem. Soc. 133 (9): 2868–2871. doi:10.1021/ja200462y. PMID 21322569.
- 1 2 Avakyan, V. G.; Sidorkin, V. F.; Belogolova, E. F.; Guselnikov, S. L.; Gusel’nikov, L. E. (2006). "AIM and ELF Electronic Structure/G2 and G3 π-Bond Energy Relationship for Doubly Bonded Silicon Species, H2SiX (X = E14H2, E15H, E16)". Organometallics. 25 (26): 6007–6013. doi:10.1021/om0605478.
- 1 2 3 Glatthaar, J.; Maier, G. (2004). "Reaktion von atomarem Silicium mit Phosphan: eine matrix‐spektroskopische Studie". Angew. Chem. 116 (10): 1314–1317. Bibcode:2004AngCh.116.1314G. doi:10.1002/ange.200353111.
- ↑ Driess, M.; Janoschek, R. (1994). "A comparison of silylidene-amines, -phosphanes and -arsanes: syntheses and quantum chemical calculations". J. Mol. Struct. 313 (1): 129–139. doi:10.1016/0166-1280(94)85036-4.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Driess, M.; Block, S.; Brym, M.; Gamer, M. T. (2006). "Synthesis of a "half"-parent phosphasilene R2Si=PH and its metalation to the corresponding P zinciophosphasilene [R2Si=PM]". Angew. Chem. Int. Ed. 45 (14): 2293–2296. doi:10.1002/anie.200504145. PMID 16518791.
- ↑ Dykema, K. J.; Truong, T. N.; Gordon, M. S. (1985). "Studies of silicon-phosphorus bonding". J. Am. Chem. Soc. 107 (15): 4535–4541. doi:10.1021/ja00301a026.
- 1 2 3 4 5 6 7 Li, B.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. (2009). "π-Conjugated Phosphasilenes Stabilized by Fused-Ring Bulky Groups". J. Am. Chem. Soc. 131 (37): 13222–13223. doi:10.1021/ja9051153. PMID 19715271.
- 1 2 3 4 5 Lee, V. Y.; Kawai, M.; Sekiguchi, A.; Ranaivonjatovo, H.; Escudié, J. (2009). "A "Push-Pull" Phosphasilene and Phosphagermene and Their Anion-Radicals". Organometallics. 28 (15): 4262–4265. doi:10.1021/om900310u.
- ↑ Ramsey, N. F. (1950). "Magnetic Shielding of Nuclei in Molecules". Phys. Rev. 78 (6): 699–703. Bibcode:1950PhRv...78..699R. doi:10.1103/PhysRev.78.699.
- 1 2 Inoue, S.; Ichinohe, M.; Sekiguchi, A. (2005). "Disilenyl Anions Derived from Reduction of Tetrakis(di-tert-butylmethylsilyl)disilene with Metal Naphthalenide through a Disilene Dianion Intermediate: Synthesis and Characterization". Chem. Lett. 34 (11): 1564–1565. doi:10.1246/cl.2005.1564.
- 1 2 Ichinohe, M.; Sanuki, K.; Inoue, S.; Sekiguchi, A. (2004). "Disilenyllithium from Tetrasila-1,3-butadiene: A Silicon Analogue of a Vinyllithium". Organometallics. 23 (13): 3088–3090. doi:10.1021/om040056s.
- 1 2 3 Hansen, K.; Szilvási, T.; Blom, B.; Irran, E. Driess, M. (2014). "A Donor-Stabilized Zwitterionic "Half-Parent" Phosphasilene and its Unusual Reactivity towards Small Molecules". Chem. Eur. J. 20 (7): 1947–1956. doi:10.1002/chem.201303906. PMID 24436015.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - 1 2 3 4 Hansen, K.; Szilvási, T.; Blom, B.; Driess, M. (2015). "A Persistent 1,2‐Dihydrophosphasilene Adduct". Angew. Chem. Int. Ed. 54 (50): 15060–15063. doi:10.1002/anie.201508149. PMID 26483284.
- ↑ Dhara, D.; Mandal, D.; Maiti, A.; Yildiz, C. B.; Kalita, P.; Chrysochos, N.; Schulzke, C.; Chandrasekhar, V.; Jana, A. (2016). "Assembly of NHC-stabilized 2-hydrophosphasilenes from Si(IV) precursors: a Lewis acid–base complex". Dalton Trans. 45 (48): 19290–19298. doi:10.1039/c6dt04321f. PMID 27872933.
- 1 2 Li, B.; Matsuo,, T.; Fukunaga, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. (2011). "Neutral and Cationic Gold(I) Complexes with π-Conjugated Phosphasilene Ligands". Organometallics. 30 (13): 3453–3456. doi:10.1021/om2003442.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - 1 2 Driess, M.; Pritzkow, H.; Winkler, U. (1997). "Synthesis of (fluorosilyl)phosphido iron and nickel complexes and ferriosilyl-substituted (fluorosilyl)phosphanes and evidence for the formation of a metallo phosphasilene of the type [M]-P=SiR2". J. Organomet. Chem. 529 (1–2): 313–321. doi:10.1016/S0022-328X(96)06483-2.
- ↑ Yao, S.; Block, S.; Brym, M.; Driess, M. (2007). "A new type of heteroleptic complex of divalent lead and synthesis of the P-plumbyleniophosphasilene, R2Si=P–Pb(L): (L = β-diketiminate)". Chem. Commun. 37 (37): 3844–3846. doi:10.1039/b710888e. PMID 18217666.
- 1 2 3 4 5 Hansen, K.; Szilvási, T.; Blom, B.; Inoue, S.; Epping, J.; Driess, M. (2013). "A Fragile Zwitterionic Phosphasilene as a Transfer Agent of the Elusive Parent Phosphinidene (:PH)". J. Am. Chem. Soc. 135 (32): 11795–11798. doi:10.1021/ja4072699. PMID 23895437.
- ↑ van den Winkel, Y.; Bastians, H. M. M.; Bickelhaupt, F. (1991). "Phosphasilene synthesis and reactivity: an improved route to 1-(2,4,6-tri-tert-butylphenyl)-2-tert-butyl-2- (2,4,6-tri-isopropylphenyl)phosphasilene". J. Organomet. Chem. 405 (2): 183–194. doi:10.1016/0022-328X(91)86271-Q.
- ↑ Scheer, M.; Balázs, G.; Seitz, A. (2010). "P4 Activation by Main Group Elements and Compounds". Chem. Rev. 110 (7): 4236–4256. doi:10.1021/cr100010e. PMID 20438122.
- ↑ Yoshifuji, M.; Toyota, K.; Ando, K.; Inamoto, N. (1984). "Isolation and Characterization of a Stable Dithioxophosphorane". Chem. Lett. 13 (3): 317–318. doi:10.1246/cl.1984.317.
- ↑ Zhu, Z.; Wang, X.; Peng, Y.; Lei, H.; Fettinger, J. C.; Rivard, E.; Power, P. P. (2009). "Addition of Hydrogen or Ammonia to a Low‐Valent Group 13 Metal Species at 25 °C and 1 Atmosphere". Angew. Chem. Int. Ed. 48 (11): 2031–2034. doi:10.1002/anie.200805982. PMID 19180617.
- ↑ Boomgaarden, S.; Saak, W.; Weidenbruch, M.; Marsmann, H. (2001). "Ammonia and Chlorine Additions to a Tetrasilabuta‐1,3‐diene: Conglomerate versus Racemate Crystallization". Z. Anorg. Allg. Chem. 627 (3): 349–352. doi:10.1002/1521-3749(200103)627:3<349::AID-ZAAC349>3.0.CO;2-R.