 | |
| Names | |
|---|---|
| Preferred IUPAC name
Phosphetane | |
| Identifiers | |
CompTox Dashboard (EPA) |
|
| Properties | |
| C3H7P | |
| Molar mass | 74.063 g·mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |
A phosphetane[1] is a 4-membered organophosphorus heterocycle. The parent phosphetane molecule, which has the formula C3H7P, is one atom larger than phosphiranes, one smaller than phospholes, and is the heavy-atom analogue of azetidines. The first known phosphetane synthesis was reported in 1957 by Kosolapoff and Struck,[2] but the method was both inefficient and hard to reproduce, with yields rarely exceeding 1%. A far more efficient method was reported in 1962 by McBride,[3] whose method allowed for the first studies into the physical and chemical properties of phosphetanes. Phosphetanes are a well understood class of molecules that have found broad applications as chemical building blocks, reagents for organic/inorganic synthesis, and ligands in coordination chemistry.
Synthesis
Many methods towards the synthesis of phosphetanes have been developed since 1957. The following are the most utilized.
McBride method (Electrophilic addition to olefins)
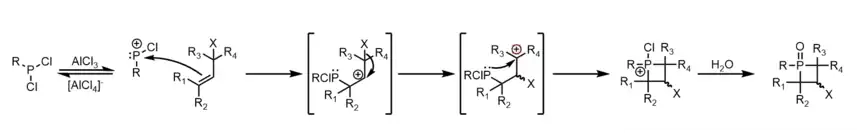
The method initially outlined by McBride has been developed for singular alkenes, as well as dienes. Both types follow the same general mechanism: formation of a phosphenium cation from a dichlorophosphine and aluminum trichloride, electrophilic addition by an alkene to the phosphenium, carbocation rearrangement, intramolecular nucleophilic addition of the new alkyl phosphine to the carbocation, and oxidation of the resulting phosphetanium with water to obtain a phosphetane oxide. Limitations of this approach are unpredictable carbocation rearrangement in more complexly branched alkanes, the incompatibility of carbocations with many nucleophilic functional groups, and the risk of cation quenching by elimination pathways.[3]
Mono-ene addition
In the case of electrophilic addition by a single alkene,[3][4] carbocation rearrangement occurs via hydride or alkyl shifts. The general scheme for phosphetane synthesis from mono-enes is given below:
Diene addition
In the case of electrophilic addition by a diene,[5][6] carbocation rearrangement first occurs via cation-π cyclization. The general scheme for phosphetane synthesis from dienes is given below:

Alkylation and intramolecular cyclization
Alkylation and cyclization pathways have been developed for both phosphines and phosphine oxides.
From phosphines
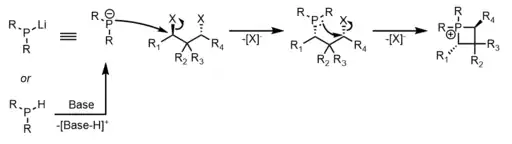
The synthesis of phosphetanes from P(III) alkylation and subsequent cyclization usually proceeds through sequential phosphanide/phosphine displacement of 1,3-alkyl dihalides or sulfonate esters (OTf, OTs, OMs, etc.).[7][8][9][10] The phosphanide source is commonly the lithium salt, but can also be accessed by in situ deprotonation of phosphines. The SN2 mechanism associated with this transformation comes with the advantage of stereospecificity, but at the expense of electrophilic or epimerizable functional group tolerance and kinetically slow reactivity with secondary/tertiary leaving groups. The general mechanism is seen below:
From phosphine oxides
Similar syntheses from P(V) compounds are known but are far rarer due to their relative inefficiency and unpredictability.[2] This preparation features the in situ formation of a Grignard reagent, followed by intramolecular addition/cyclization to a phosphine oxide, all on an n-propyl backbone. This was the method employed by Kosolapoff and Struck in the first synthesis of a phosphetane. The general mechanism is seen below:

Cyclopropane ring-expansion
Another way to make phosphetanes comes from the ring-expansion of cyclopropanes, in which it seems a phosphine is directly inserted into a C-C bond.[11][12] The true mechanism of this transformation is similar to that of the McBride synthesis and is sometimes classified as such, with similar advantages and drawbacks. Although relieving the cyclopropane ring strain is of great assistance in the initial C-P bond, exhaustive alkyl substitution to stabilize the formed carbocation is often required. The general mechanism is seen below:

[2+2] cycloaddition
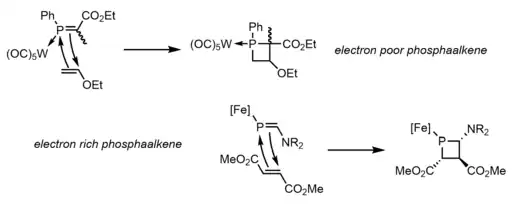
One final method that has been observed to produce phosphetanes is the [2+2] cycloaddition of phosphaalkenes and olefins. This method is not often discussed for its tendency to produce phosphetanes, but rather for its insight into the reactivity of the much more elusive phosphaalkenes. The difficult synthesis of these phosphaalkenes severely limits the utility of the method as it relates to phosphetane synthesis, despite its attractive stereospecific and modular approach. This usually involves a Lewis acid bound phosphorus, and can occur with electron rich phosphaalkenes and electron poor olefins,[13] or the inverse.[14] An example of each, and the mechanism, are seen below:
Structure and bonding
Experimental and crystallographic data exists for many of the phosphetane types listed below, however, all of the geometric and electronic (HOMO and LUMO) information below was determined theoretically with the B3LYP functional[15][16][17][18] and DEF2-SVP basis set[19] using ORCA (5.0.3)[20] for the parent molecule at each coordination number to provide a general and consistent trend as an introduction to the subject. Geometries and orbital plots were generated using Avogadro (4.1).[21]
Dicoordinate phosphetanes
Though rarely reported in the literature, if at all, dicoordinate phosphetanes of phosphenium, phosphanide, and phosphorus radical archetypes are theoretically possible as transient reactive intermediates. Their optimized physical and electronic geometries are presented mainly as a means of comparison to the more commonly observed tri, tetra, and pentacoordinate phosphetanes.
Phosphenium ion
The phosphenium case is isoelectronic to a cyclic carbene. The optimized geometry is quite planar in comparison to the other dicoordinate cases, with its HOMO and LUMO being the exocyclic lone pair and empty p-orbital, respectively.
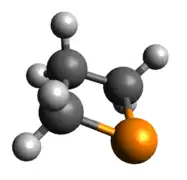 Optimized Geometry
Optimized Geometry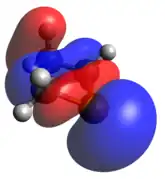 HOMO
HOMO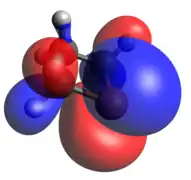 LUMO
LUMO
Phosphorus radical
The optimized geometry and frontier molecular orbitals for the dicoordinate phosphorus radical are similar to the phosphenium case. The ring is slightly less planar, and the HOMO is now a singly occupied p-orbital. The lone pair is the HOMO-1.
 Optimized Geometry
Optimized Geometry HOMO
HOMO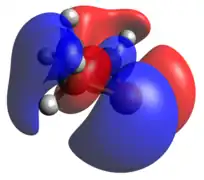 LUMO
LUMO
Phosphanide ion
The phosphanide case is isoelectronic to cyclic ethers. In this ion, there is significantly more pucker within the phosphetane ring, along with widening of the C-P-C angle, but the HOMO and HOMO-1 are similar to the radical case, now both being doubly occupied.
 Optimized Geometry
Optimized Geometry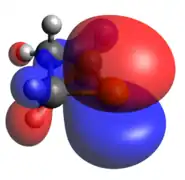 HOMO
HOMO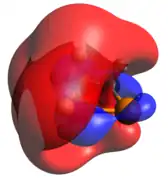 LUMO
LUMO
Tricoordinate phosphetanes
Tricoordinate phosphetanes are well known in the literature and exemplify the classical trigonal pyramidal P(III) phosphorus center. Conformational isomerism is introduced in these tricoordinate molecules, albeit with a very low kinetic barrier (~2.45 kcal/mol for the given example),[22] in which the hydrogen can be pseudo-axial (as shown), or pseudo-equatorial. The pseudo-axial conformer is the more stable of the two. Since the lone pair is larger, it settles in the pseudo-equatorial position, but this is inverted rather swiftly due to minimization of steric clash as R becomes bigger than H. The phosphetane ring is puckered, not planar, due to the asymmetry above and below the ring about phosphorus. As is expected, the HOMO is the nucleophilic lone pair usually associated with phosphines.
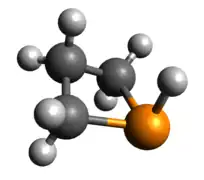 Optimized Geometry
Optimized Geometry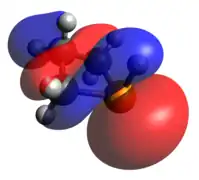 HOMO
HOMO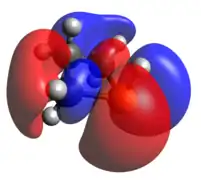 LUMO
LUMO
Tetracoordinate phosphetanes
Tetracoordinate phosphetanes are by far the most commonly observed geometry around the phosphorus center, usually as the ubiquitous P(V) phosphorus oxide center, but not uncommonly as phosphetanium ions.
Phosphetanium ion
The phosphetanium is isoelectronic to a tetracoordinate carbon and assumes its tetrahedral geometry, greatly planarizing the ring by increasing molecular symmetry. Deviation from this would occur with any change of one of the hydrogen atoms with a bulkier group, after which, the ring would pucker, with the larger substituent pseudo-equatorial. The acidity of the α-carbon hydrogens is significantly increased due to the charge neutralization driving force; this is reflected in the C-H σ-antibonding contributions to the LUMO.
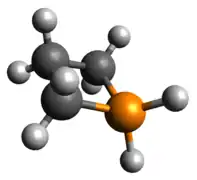 Optimized Geometry
Optimized Geometry HOMO
HOMO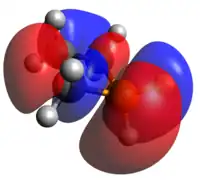 LUMO
LUMO
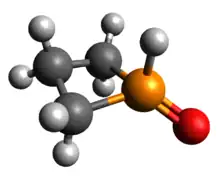
Phosphetane oxide
The other classic phosphorus compound is the tetrahedral P(V) phosphine oxide. Like tricoordinate phosphetanes, phosphetane oxides also exhibit isomerism, this time with a much larger kinetic barrier. When the oxide is pseudo-equatorial (as shown), the designation is trans, while when the oxide is pseudo-axial, the compound is cis. The preference for one over the other is largely based on the middle carbon substitution, rather than the oxide.[23] As one may expect of a covalently bound oxide, the HOMO is an oxygen lone pair and the LUMO is largely contributed to by the P-O π-antibonding interaction.
 Optimized Geometry
Optimized Geometry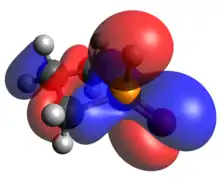 HOMO
HOMO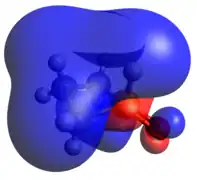 LUMO
LUMO
Pentacoordinate phosphetanes
Pentacoordinate phosphetanes, or phosphoranes, present an alternative geometric mantle on which a P(V) phosphorus center may exist. It is important to note that this class of phosphoranes are typically not trigonal bipyramidal, but closer to square pyramidal. A result of this geometric perturbation is the emergence of a P-H σ-antibonding that is represented prominently in the LUMO, accounting for the characteristic Lewis acidity of square pyramidal phosphoranes.
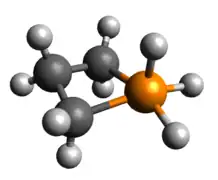 Optimized Geometry
Optimized Geometry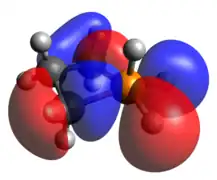 HOMO
HOMO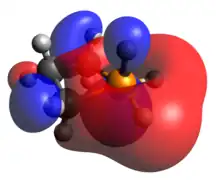 LUMO
LUMO
Hexacoordinate phosphetanes
Hexacoordinate, anionic phosphates are mainly known in the literature as counterions (hexafluorophosphate), but are theoretically possible as reactive intermediates for associative mechanisms at phosphorus centers. In this compound, phosphorus assumes the expected octahedral geometry. As expected for this hexacoordinate phosphate, C-H σ-bonding orbitals comprise the HOMO, accounting for the expected hydricity due to favorable charge neutralization. Similar to the dicoordinate case, these optimized physical and electronic geometries are presented mainly as a means of comparison to the more commonly observed tri, tetra, and pentacoordinate phosphetanes.
 Optimized Geometry
Optimized Geometry HOMO
HOMO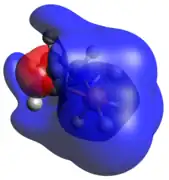 LUMO
LUMO
Reactivity
Phoshetanes display a broad range of reactivity and appear in the literature in many different facets of a chemical reaction. There are cases where phosphetanes themselves are the substrate of interest, cases where phosphetanes are observed as transient intermediates during a chemical reaction, cases where phosphetanes are used as the active reagents in chemical reactions, and cases where phosphetanes are ligated to a metal that is the active reagent in a given process. All of these overarching scenarios are discussed in more detail below.
Inherent reactivity
Much of the reactivity inherent to, or performed directly on, phosphetane substrates is an ode to its ring strain, calculated to be ~17.9 kcal/mol.[22] The release of some or all of this strain energy drive the two characteristic types of reactivity observed: ring expansion and ring opening. Reactivity at the phosphorus center, including reduction, oxidation, and phosphorane formation as well as alkylation of ring carbons can be performed without cleavage of the ring in some instances, representing the final types of inherent reactivity. These four will be discussed in more detail below.
Ring opening reactions
Phosphetane ring opening reactions have been of synthetic interest in the past as a potential method for the creation of polypropylphosphine polymers and materials, but despite ring opening reactions occurring, the polymerization of such material has only been sparsely observed in very concentrated solutions.[8][24]
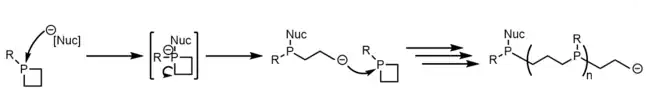
The main observation of ring opening is as a byproduct of other intended reactions, such as phosphetanium oxidation[25][26] and α-carbon functionalization.[27]

One intentional and constructive method of ring-opening has been outlined in the literature and features a phosphetane ylide that undergoes Wittig reactivity with aldehydes to form γ-unsaturated phosphine oxides.[28]

Ring expansion reactions
Methods of ring expansion to insert carbon, oxygen, and nitrogen atoms into phosphetane rings to produce the corresponding phospholes exist but are of limited synthetic utility due to their unpredictable stereo and regioselectivity on unsymmetric phosphetanes. Insertion of carbon typically involves the addition of water to a phosphetanium featuring a leaving group[25] or pi-system[29][25] (usually enones but also phenyl groups) alpha to phosphorus that is liberated by alkyl migration after collapse of the phosphetane oxide.
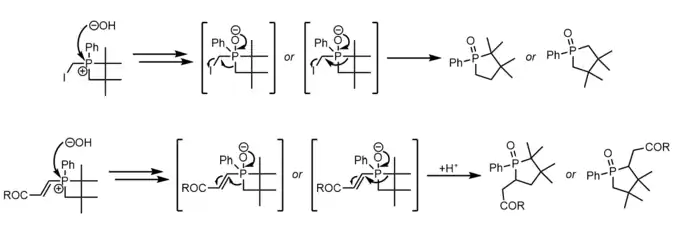
Insertion of oxygen into the P-C bond of a phosphetane oxide is done with mCPBA and proceeds via a currently unknown mechanism with unusually high regioselectivity for the less substituted carbon.[30][31]

Nitrogen atom insertion proceeds from photolysis of an azidophosphetane oxide, presumably from a Curtius type rearrangement from the generated nitrene. Though this is the proposed mechanism, there are clear doubts about the N=P=O intermediate.[32][33]

Reactivity at phosphorus
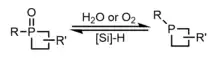
Redox between P(III) phosphetanes and P(V) phosphetane oxides are possible and well documented through the use of mild reagents such as oxygen or water and silicon hydrides to achieve oxidation and reduction, respectively.
More interesting is the synthesis of stable 5-coordinate phosphetanes (phosphoranes) from both traditional P(III) phosphines and P(V) phosphine oxides, in addition to P(V) phosphetanium ions, via a couple general methods. With respect to phosphine substrates, phosphorane synthesis usually occurs via reaction with peroxides/disulfides[34][35][36] or perfluoro π-systems, such as perfluoro acetone,[37] for which the mechanism is unresolved, or perfluoro 1,3-butadiene.[38]
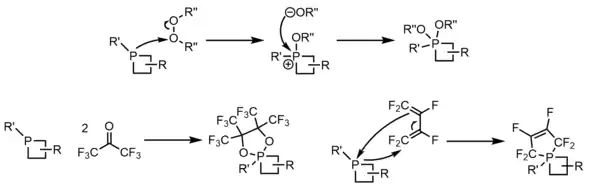
Methods to access phosphoranes from P(V) oxides and phosphetaniums are usually through stepwise deoxygenation-nucleophilic addition pathways,[39] or direct addition pathways,[28] respectively. Nucleophiles are usually halides or alkoxy functional groups, and in the case of deoxygenation-substitution, the two nucleophiles can be either tethered (e.g. catechol) or not.
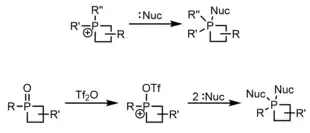
α-Carbon functionalization
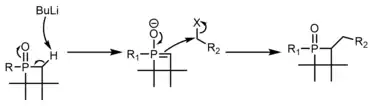
The final portion of inherent reactivity of phosphetanes to be discussed is the functionalization of the phosphetane oxide alpha carbons, almost always through deprotonation with organolithium reagents, followed by SN2 displacement of an alkyl halide.[40] The use of chiral axillaries on phosphorus can make this process stereoselective.[41][42]
Reactive intermediates
The appearance of phosphetanes and derivatives thereof is well documented in organic chemistry literature as reactive intermediates for a myriad of different processes. These processes include, but are not limited to, Wittig, Horner-Wadsworth-Emmons, Corey-Fuchs, and Seyferth-Gilbert chemistries. All of these processes include the in-situ formation and decomposition of oxaphosphetane intermediates through metathesis-type pathways to form alkenes or alkynes from aldehydes and a phosphorus reagent.
Reagents and catalysts
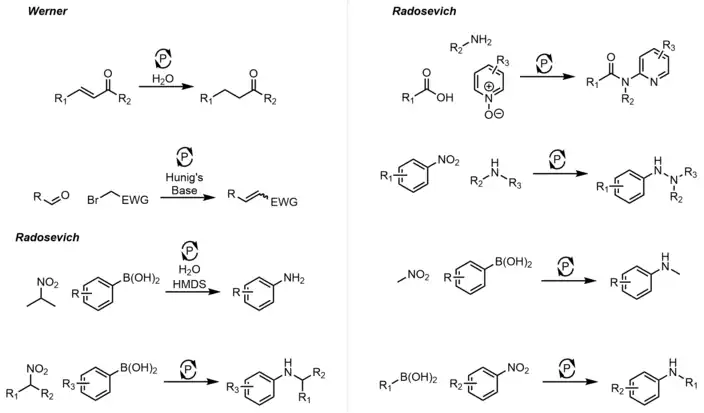
Since the early 2010s, much progress has been made in the development of phosphetanes as useful reagents and catalysts to complement transition metal catalysts in organic synthesis. These efforts have primarily been made by the research group of Dr. Alexander Radosevich[43][44][45][46][47][48] at Pennsylvania State University, and subsequently the Massachusetts Institute of Technology, but contributions from the lab of Dr. Thomas Werner[49][50] at the Leibniz-Institut für Katalyse (Leibniz Institute for Catalysis) have also been impactful. The common theme underpinning these works is an active phosphetane species reductively acting on a substrate, resulting in formation of phosphetane oxide and the desired product, followed by reduction of the phosphetane oxide back to the phosphetane with a mild silicon hydride which closes the catalytic cycle.
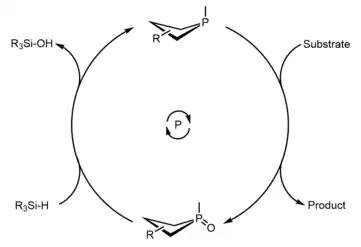
The uncharacteristic biphilic nature of these phosphines, and other non-trigonal pnictogen compounds, is a result of molecular symmetry perturbation,[51] in this case, imposed by the ring strain inherent to phospetanes. Most of these transformations are probed based on stoichiometric reactivity of the phosphetane, illustrating their utility as catalysts or reagents in the event there is substrate incompatibility with the hydride. Below is the general catalytic cycle and an abbreviated list of reactions that can be catalyzed through this method.
Ligands for transition metal complexes
Transition metal complexes with ligated P(III) phosphetanes are known for tungsten,[52] iron,[53][54][13] molybdenum,[55][8] platinum,[24] ruthenium,[56][57] rhodium,[58][57][59] palladium,[42][56][60] iridium,[42] and possibly more, to produce achiral, racemic, and optically pure coordination complexes. Despite these efforts, the intricate details about their nature as ligands and effects on metal centers as it deviates from traditional phosphines is relatively understudied. Direct comparison of classic bis-trialkylphosphinedichloroplatinum(II) complexes with the corresponding phosphetane containing complex possibly enumerate a weakened σ-trans effect and π-accepting character of the phosphetane ligand, most likely due to the aforementioned symmetry distortion, corroborated by short Pt-P (2.208 and 2.210 angstrom) and Pt-Cl (2.342 and 2.355 angstrom) bonds.[24] More work is needed to make this claim categorically.
Most of the study and interest in phosphetanes as ligands is there ability to impart enantioselectivity on certain catalytic hydrogenation,[61] reduction,[60] and π-allyl[62] reactions when using the corresponding chiral phosphetanes. As is the case for most asymmetric catalysis, disfavored steric interaction between chiral ligands, substrate, and other reagents are credited for the observed enantio or diastereoselectivity, though it seems the use of more traditional chiral phosphines has proved more popular than that of chiral phosphetanes. Below are select examples of enantioselective catalysis using phosphetane ligands.
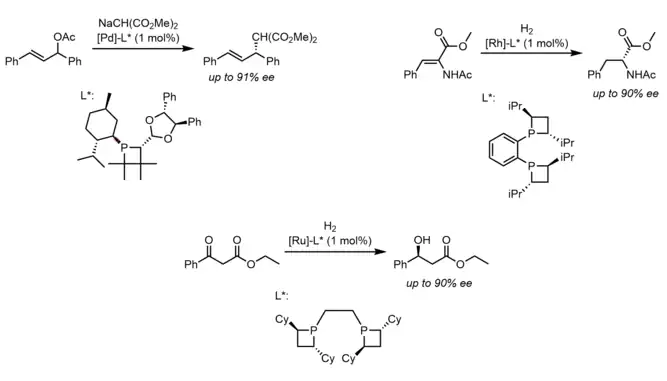
References
- ↑ Marinetti, Angela; Carmichael, Duncan (2002-01-01). "Synthesis and Properties of Phosphetanes". Chemical Reviews. 102 (1): 201–230. doi:10.1021/cr990135r. ISSN 0009-2665. PMID 11782133.
- 1 2 Kosolapoff, Gennady M.; Struck, Robert F. (1957-01-01). "736. The dissociation constants of some cyclic phosphinic acids". Journal of the Chemical Society (Resumed): 3739–3740. doi:10.1039/JR9570003739. ISSN 0368-1769.
- 1 2 3 Jungermann, Eric; McBride, J. J.; Clutter, R.; Mais, Ago (February 1962). "A New Phosphorylation Reaction of Olefins. I. Scope of the Reaction". The Journal of Organic Chemistry. 27 (2): 606–610. doi:10.1021/jo01049a063. ISSN 0022-3263.
- ↑ Vilkas, Erna; Vilkas, Michel; Joniaux, Denise; Pascard-Billy, Claudine (1978-01-01). "1-Isopropyl-4,6-dimethyl-6-phospha(V)bicyclo[3.1.1]hept-3-ene 6-oxide: a distorted phosphetan synthesized from α-pinene and the methylphosphonous dichloride–aluminium chloride complex; X-ray crystal and molecular structure". Journal of the Chemical Society, Chemical Communications (3): 125–126. doi:10.1039/C39780000125. ISSN 0022-4936.
- ↑ Weissman, Steven A.; Baxter, S. G.; Arif, Atta M.; Cowley, Alan H. (1986-01-01). "Reaction of a halogenophosphenium ion with cyclo-octa-1,5-diene; direct synthesis and X-ray crystal structure of a phosphetane moiety". Journal of the Chemical Society, Chemical Communications (14): 1081–1082. doi:10.1039/C39860001081. ISSN 0022-4936.
- ↑ Cowley, Alan H.; Stewart, Constantine A.; Whittlesey, Bruce R.; Wright, Thomas C. (1984-01-01). "Phosphorus heterocycle synthesis using phosphenium ions and 1,4-dienes". Tetrahedron Letters. 25 (8): 815–816. doi:10.1016/S0040-4039(01)80034-4. ISSN 0040-4039.
- ↑ Berglund, Donna; Meek, Devon W. (January 1968). "Coordination by a positively charged phosphorus ligand". Journal of the American Chemical Society. 90 (2): 518–519. doi:10.1021/ja01004a071. ISSN 0002-7863.
- 1 2 3 Hockless, David C. R.; Kang, Yew Beng; McDonald, Mark A.; Pabel, Michael; Willis, Anthony C.; Wild, S. Bruce (1996-02-20). "Direct Syntheses of 1-Phenylphosphetane and 1-Phenylphosphirane. Crystal and Molecular Structures of Neutral and Cationic Cyclotrimerization Precursor Complexes". Organometallics. 15 (4): 1301–1306. doi:10.1021/om9507336. ISSN 0276-7333.
- ↑ Marinetti, Angela; Kruger, Virginie; Buzin, François-Xavier (1997-04-28). "Synthesis of Chiral Phosphetanes". Tetrahedron Letters. 38 (17): 2947–2950. doi:10.1016/S0040-4039(97)00508-X. ISSN 0040-4039.
- ↑ Imamoto, Tsuneo; Ohashi, Atsushi; Matsukawa, Satoru (2000). "Improved Synthesis of 1-Adamantylphosphine and Its Use in the Synthesis of Cyclicphosphines Containing 1-Adamantyl Group". Heterocycles. 52 (2): 905. doi:10.3987/COM-99-S58. ISSN 0385-5414.
- ↑ Weissman, Steven A.; Baxter, S. G. (1988-01-01). "Insertion of electrophilic phosphorus into cyclopropanes; a new synthesis of phosphatanes". Tetrahedron Letters. 29 (11): 1219–1222. doi:10.1016/S0040-4039(00)80260-9. ISSN 0040-4039.
- ↑ Weissman, Steven A.; Baxter, S. G. (1987-01-01). "Cycloaddition of phosphenium ions to norbornadiene and quadricyclane". Tetrahedron Letters. 28 (6): 603–606. doi:10.1016/S0040-4039(00)95791-5. ISSN 0040-4039.
- 1 2 Weber, Lothar; Scheffer, Matthias H.; Beckmann, Eike; Stammler, Hans-Georg; Neumann, Beate (1997-06-01). "Transition-Metal-Substituted Acyl Phosphanes and Phosphaalkenes. 32. 1 Transformation of an FePC Unit of a Phosphaalkene into a FeP C N N C Moiety by Chain Extension through Insertion of the 1,3-Dipole ( - NN(Ar)CH + ) of Sydnones". Organometallics. 16 (13): 2958–2962. doi:10.1021/om9700876. ISSN 0276-7333.
- ↑ Marinetti, Angela; Mathey, François (1990-01-01). "[2 + 2] Cycloadditions between electron-poor phospha-alkene complexes and electron-rich alkenes or alkynes, a new route to phosphetane and 1,2-dihydrophosphete rings". Journal of the Chemical Society, Chemical Communications (2): 153–154. doi:10.1039/C39900000153. ISSN 0022-4936.
- ↑ Becke, Axel D. (1993-04-01). "Density‐functional thermochemistry. III. The role of exact exchange". The Journal of Chemical Physics. 98 (7): 5648–5652. Bibcode:1993JChPh..98.5648B. doi:10.1063/1.464913. ISSN 0021-9606. S2CID 52389061.
- ↑ Lee, Chengteh; Yang, Weitao; Parr, Robert G. (1988-01-15). "Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density". Physical Review B. 37 (2): 785–789. Bibcode:1988PhRvB..37..785L. doi:10.1103/PhysRevB.37.785. PMID 9944570.
- ↑ Vosko, S. H.; Wilk, L.; Nusair, M. (1980-08-01). "Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis". Canadian Journal of Physics. 58 (8): 1200–1211. Bibcode:1980CaJPh..58.1200V. doi:10.1139/p80-159. ISSN 0008-4204. S2CID 122287164.
- ↑ Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. (November 1994). "Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields". The Journal of Physical Chemistry. 98 (45): 11623–11627. doi:10.1021/j100096a001. ISSN 0022-3654. S2CID 97035345.
- ↑ Weigend, Florian; Ahlrichs, Reinhart (2005-08-30). "Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy". Physical Chemistry Chemical Physics. 7 (18): 3297–3305. Bibcode:2005PCCP....7.3297W. doi:10.1039/B508541A. ISSN 1463-9084. PMID 16240044.
- ↑ Neese, Frank (January 2012). "The ORCA program system". WIREs Computational Molecular Science. 2 (1): 73–78. doi:10.1002/wcms.81. ISSN 1759-0876. S2CID 62137389.
- ↑ Hanwell, Marcus D.; Curtis, Donald E.; Lonie, David C.; Vandermeersch, Tim; Zurek, Eva; Hutchison, Geoffrey R. (2012-08-13). "Avogadro: an advanced semantic chemical editor, visualization, and analysis platform". Journal of Cheminformatics. 4 (1): 17. doi:10.1186/1758-2946-4-17. ISSN 1758-2946. PMC 3542060. PMID 22889332.
- 1 2 Bachrach, Steven M. (November 1989). "Theoretical studies of phosphirane and phosphetane". The Journal of Physical Chemistry. 93 (23): 7780–7784. doi:10.1021/j100360a011. ISSN 0022-3654.
- ↑ Campbell, J. A.; Caughlan, C. N.; Fitzgerald, A.; Campana, C.; Cremer, S. E. (1984-11-15). "The structure of 1-tert-butyl-2,2,3,4,4-pentamethylphosphetane 1-oxide, C12H25OP". Acta Crystallographica Section C: Crystal Structure Communications. 40 (11): 1918–1920. doi:10.1107/S0108270184010052. ISSN 0108-2701.
- 1 2 3 Tumas, William; Huang, Jean C.; Fanwick, Phillip E.; Kubiak, Clifford P. (August 1992). "Synthesis and reactivity of cyclic phosphetanes. Oligomerization, quaternization, and complexation with platinum(II)". Organometallics. 11 (8): 2944–2947. doi:10.1021/om00044a042. ISSN 0276-7333.
- 1 2 3 Corfield, J. R.; Harger, M. J. P.; Shutt, J. R.; Trippett, S. (1970-01-01). "Further ring openings and ring expansions of phosphetans". Journal of the Chemical Society C: Organic (13): 1855–1860. doi:10.1039/J39700001855. ISSN 0022-4952.
- ↑ Ezzell, B. R. (July 1970). "Alkaline cleavage of phosphetane oxides". The Journal of Organic Chemistry. 35 (7): 2426–2428. doi:10.1021/jo00832a079. ISSN 0022-3263.
- ↑ Cremer, Sheldon E.; Cowles, John M.; Farr, Frank R.; Hwang, Hai Ok; Kremer, Paul W.; Peterson, Andrew C.; Gray, George A. (January 1992). "Preparation, reactions, and stereochemistry of 4-methyl-4-phosphatetracyclo[3.3.0.02,8.03,6]octane 4-oxide and derivatives". The Journal of Organic Chemistry. 57 (2): 511–522. doi:10.1021/jo00028a022. ISSN 0022-3263.
- 1 2 Brauer, David J.; Ciccu, Antonella J.; Heßler, Gisbert; Stelzer, Othmar (September 1992). "Neue Synthesen und Reaktionen von Phosphetaniumsalzen mit planarem Gerüst". Chemische Berichte. 125 (9): 1987–1997. doi:10.1002/cber.19921250904. ISSN 0009-2940.
- ↑ Hawes, W.; Trippett, S. (1969-01-01). "Some substitution reactions at the phosphorus of 2,2,3,4,4-pentamethyl-phosphetans". Journal of the Chemical Society C: Organic (11): 1465–1468. doi:10.1039/J39690001465. ISSN 0022-4952.
- ↑ Quin, Louis D.; Kisalus, John C.; Mesch, Keith A. (December 1983). "Insertion of oxygen into carbon-phosphorus bonds of some strained phosphorus heterocycles". The Journal of Organic Chemistry. 48 (24): 4466–4472. doi:10.1021/jo00172a005. ISSN 0022-3263.
- ↑ Szewczyk, Jerzy; And, En-Yun Yao; Quin, Louis D. (September 1990). "Synthesis of 1,2-Oxaphspholane Oxides By Oxgen Insertion Into the C-P Bond of Phosphetane Oxides". Phosphorus, Sulfur, and Silicon and the Related Elements. 54 (1–4): 135–141. doi:10.1080/10426509008042130. ISSN 1042-6507.
- ↑ Wiseman, Jeffrey; Westheimer, F. H. (June 1974). "Photolysis of 1-azido-2,2,3,4,4-pentamethylphosphetane 1-oxide. Monomeric metaphosphonimidate". Journal of the American Chemical Society. 96 (13): 4262–4268. doi:10.1021/ja00820a033. ISSN 0002-7863.
- ↑ Harger, Martin J. P. (1974-01-01). "Photolysis of 1-azidophosphetan oxides: ring expansion to 2-methoxy-1,2-azaphospholidine 2-oxides and ring opening to methyl alk–ylphosphonamidates in methanol". Journal of the Chemical Society, Perkin Transactions 1: 2604–2609. doi:10.1039/P19740002604. ISSN 1364-5463.
- ↑ Denney, Donald B.; Denney, Dorothy Z.; White, Dennis W. (April 1971). "Remarkably facile pseudorotation of four-membered-ring phosphoranes". Journal of the American Chemical Society. 93 (8): 2066–2067. doi:10.1021/ja00737a047. ISSN 0002-7863.
- ↑ Denney, D. B.; Denney, D. Z.; Hall, C. D.; Marsi, K. L. (January 1972). "Preparation and chemistry of some cyclic phosphoranes". Journal of the American Chemical Society. 94 (1): 245–249. doi:10.1021/ja00756a043. ISSN 0002-7863.
- ↑ Burros, Byron C.; De'Ath, Norman J.; Denney, Donald B.; Denney, Dorothy Z.; Kipnis, Irving J. (November 1978). "Preparation and chemistry of some phosphoranes containing phosphorus-sulfur bonds". Journal of the American Chemical Society. 100 (23): 7300–7304. doi:10.1021/ja00491a029. ISSN 0002-7863.
- ↑ Oram, Robert K.; Trippett, Stuart (1973-01-01). "Reactions of 1-substituted 2,2,3,4,4-pentamethylphosphetans with hexafluoroacetone and the 19F nuclear magnetic resonance spectra of the resulting 1,3,2-dioxaphospholans". Journal of the Chemical Society, Perkin Transactions 1: 1300–1310. doi:10.1039/P19730001300. ISSN 1364-5463.
- ↑ Denney, D. B.; Denney, D. Z.; Hsu, Y. F. Phosphorus, 1974, 4, 217.
- ↑ Antczak, Stephen; Trippett, Stuart (1978-01-01). "Synthesis of quinquevalent phosphoranes from phosphine oxides". Journal of the Chemical Society, Perkin Transactions 1 (11): 1326–1330. doi:10.1039/P19780001326. ISSN 1364-5463.
- ↑ Marinetti, Angela; Ricard, Louis (1993-01-01). "Synthesis and characterization of some p-methylphosphetanes, a new class of electron-rich chiral phosphines". Tetrahedron. 49 (45): 10291–10304. doi:10.1016/S0040-4020(01)80558-5. ISSN 0040-4020.
- ↑ Marinetti, Angela; Buzin, François-Xavier; Ricard, Louis (1997-03-24). "Synthesis and α-alkylation of 1-menthylphosphetane sulfide". Tetrahedron. 53 (12): 4363–4370. doi:10.1016/S0040-4020(97)00132-4. ISSN 0040-4020.
- 1 2 3 Marinetti, Angela; Ricard, Louis (October 1994). "Phosphetanes as Chiral Ligands for Catalytic Asymmetric Reactions: Hydrosilylation of Olefins". Organometallics. 13 (10): 3956–3962. doi:10.1021/om00022a035. ISSN 0276-7333.
- ↑ Hong, Seung Youn; Radosevich, Alexander T. (2022-05-25). "Chemoselective Primary Amination of Aryl Boronic Acids by P III /P V ═O-Catalysis: Synthetic Capture of the Transient Nef Intermediate HNO". Journal of the American Chemical Society. 144 (20): 8902–8907. doi:10.1021/jacs.2c02922. ISSN 0002-7863. PMC 9133210. PMID 35549268.
- ↑ Li, Gen; Kanda, Yuzuru; Hong, Seung Youn; Radosevich, Alexander T. (2022-05-11). "Enabling Reductive C–N Cross-Coupling of Nitroalkanes and Boronic Acids by Steric Design of P(III)/P(V)═O Catalysts". Journal of the American Chemical Society. 144 (18): 8242–8248. doi:10.1021/jacs.2c01487. ISSN 0002-7863. PMC 9119554. PMID 35499970.
- ↑ Lipshultz, Jeffrey M.; Radosevich, Alexander T. (2021-09-15). "Uniting Amide Synthesis and Activation by P III /P V –Catalyzed Serial Condensation: Three-Component Assembly of 2-Amidopyridines". Journal of the American Chemical Society. 143 (36): 14487–14494. doi:10.1021/jacs.1c07608. ISSN 0002-7863. PMC 9088293. PMID 34478308.
- ↑ Li, Gen; Miller, Steven P.; Radosevich, Alexander T. (2021-09-15). "P III /P V ═O-Catalyzed Intermolecular N–N Bond Formation: Cross-Selective Reductive Coupling of Nitroarenes and Anilines". Journal of the American Chemical Society. 143 (36): 14464–14469. doi:10.1021/jacs.1c07272. ISSN 0002-7863. PMC 8454687. PMID 34473484.
- ↑ Li, Gen; Qin, Ziyang; Radosevich, Alexander T. (2020-09-23). "P(III)/P(V)-Catalyzed Methylamination of Arylboronic Acids and Esters: Reductive C–N Coupling with Nitromethane as a Methylamine Surrogate". Journal of the American Chemical Society. 142 (38): 16205–16210. doi:10.1021/jacs.0c08035. ISSN 0002-7863. PMC 7531042. PMID 32886500.
- ↑ Nykaza, Trevor V.; Cooper, Julian C.; Li, Gen; Mahieu, Nolwenn; Ramirez, Antonio; Luzung, Michael R.; Radosevich, Alexander T. (2018-11-14). "Intermolecular Reductive C–N Cross Coupling of Nitroarenes and Boronic Acids by P III /P V ═O Catalysis". Journal of the American Chemical Society. 140 (45): 15200–15205. doi:10.1021/jacs.8b10769. ISSN 0002-7863. PMC 6235741. PMID 30372615.
- ↑ Longwitz, Lars; Werner, Thomas (2020-02-10). "Reduction of Activated Alkenes by P III /P V Redox Cycling Catalysis". Angewandte Chemie International Edition. 59 (7): 2760–2763. doi:10.1002/anie.201912991. ISSN 1433-7851. PMC 7027467. PMID 31793147.
- ↑ Longwitz, Lars; Spannenberg, Anke; Werner, Thomas (2019-10-04). "Phosphetane Oxides as Redox Cycling Catalysts in the Catalytic Wittig Reaction at Room Temperature". ACS Catalysis. 9 (10): 9237–9244. doi:10.1021/acscatal.9b02456. ISSN 2155-5435. S2CID 202880682.
- ↑ Lee, Kyounghoon; Blake, Anastasia V.; Tanushi, Akira; McCarthy, Sean M.; Kim, Daniel; Loria, Sydney M.; Donahue, Courtney M.; Spielvogel, Kyle D.; Keith, Jason M.; Daly, Scott R.; Radosevich, Alexander T. (2019-05-20). "Validating the Biphilic Hypothesis of Nontrigonal Phosphorus(III) Compounds". Angewandte Chemie International Edition. 58 (21): 6993–6998. doi:10.1002/anie.201901779. ISSN 1433-7851. PMC 6513703. PMID 30901511.
- ↑ Rohde, Udo; Ruthe, Frank; Jones, Peter; Streubel, Rainer (January 18, 1999). "Formation and Structure of the First 7-Aza-1-phosphanorbornadiene Complex". Angewandte Chemie International Edition. 38 (1–2): 215–217. doi:10.1002/(SICI)1521-3773(19990115)38:1/2<215::AID-ANIE215>3.0.CO;2-Y.
- ↑ Bader, Armin; Pathak, Devendra D.; Wild, S. Bruce; Willis, Anthony C. (1992-01-01). "Dalton communications. Reactions of co-ordinated phosphines and arsines. Iron(II)-facilitated syntheses of 1-phenylphosphetane and 1-phenylarsetane". Journal of the Chemical Society, Dalton Transactions (10): 1751–1752. doi:10.1039/DT9920001751. ISSN 1364-5447.
- ↑ Bader, Armin; Kang, Yew Beng; Pabel, Michael; Pathak, Devendra D.; Willis, Anthony C.; Wild, S. Bruce (March 1995). "Reactions of Coordinated Phosphines and Arsines. Iron(II)-Facilitated and Direct Syntheses of Three- to Seven-Membered Heterocycles Containing Phosphorus and Arsenic. Crystal Structures of Iron(II) Complexes of 1-Phenylphosphetane and 1-Phenylarsetane". Organometallics. 14 (3): 1434–1441. doi:10.1021/om00003a051. ISSN 0276-7333.
- ↑ Kang, Yew Beng; Pabel, Michael; Willis, Anthony C.; Wild, S. Bruce (1994-01-01). "Direct syntheses of 1-phenylphosphetane and 1-phenylphosphirane. Crystal and molecular structures of cyclotrimerisation precursor complexes fac-[Mo(CO)3(PhPCH2CH2CH2)3] and fac-[Mo(CO)3(PhPCH2CH2)3]". Journal of the Chemical Society, Chemical Communications (4): 475–476. doi:10.1039/C39940000475. ISSN 0022-4936.
- 1 2 Marinetti, Angela; Jus, Sébastien; Genêt, Jean-Pierre; Ricard, Louis (2000-01-01). "Additional Data on the Synthesis and Properties of Chiral 1,2-Bis(phosphetano)benzenes". Tetrahedron. 56 (1): 95–100. doi:10.1016/S0040-4020(99)00777-2. ISSN 0040-4020.
- 1 2 Marinetti, A. (October 1998). "Unsymmetrical Bidentate Ligands Bearing Chiral Phosphetane Units". Synthesis. 1998 (10): 1539–1543. doi:10.1055/s-1998-2169.
- ↑ Marinetti, Angela; Jus, Sébastien; Labrue, Francis; Lemarchand, Aude; Genêt, Jean-Pierre; Ricard, Louis (2001). "Synthesis and Characterisation of Monophosphines and Aminophosphines Bearing Chiral Phosphetane Units". Synthesis. 2001 (14): 2095–2104. doi:10.1055/s-2001-18064.
- ↑ Marinetti, Angela; Le Menn, Claude; Ricard, Louis (November 1995). "Mono- and Binuclear Rhodium Complexes of a Chiral 1,1-Diphosphine. Syntheses and Crystal Structures". Organometallics. 14 (11): 4983–4985. doi:10.1021/om00011a006. ISSN 0276-7333.
- 1 2 Marinetti, Angela; Jus, Sébastien; Genêt, Jean-Pierre; Ricard, Louis (2001-04-01). "Chiral 1,2-bis(phosphetano)ethanes". Journal of Organometallic Chemistry. 624 (1): 162–166. doi:10.1016/S0022-328X(00)00910-4. ISSN 0022-328X.
- ↑ Marinetti, Angela; Jus, Sébastien; Genêt, Jean-Pierre (1999-11-26). "Investigation into an asymmetric hydrogenation promoted by rhodium-phosphetane complexes". Tetrahedron Letters. 40 (48): 8365–8368. doi:10.1016/S0040-4039(99)01767-0. ISSN 0040-4039.
- ↑ Marinetti, Angela; Kruger, Virginie; Ricard, Louis (1997-02-15). "New chiral phosphetanes: Synthesis and use in the palladium-catalyzed allylic alkylation". Journal of Organometallic Chemistry. 529 (1): 465–472. doi:10.1016/S0022-328X(96)06563-1. ISSN 0022-328X.