Phosphorus-centered (or P-centered) porphyrins are conjugated polycyclic ring systems consisting of either four pyrroles with inward-facing nitrogens and a phosphorus atom at their core or porphyrins with one of the four pyrroles substituted for a phosphole. Unmodified porphyrins are composed of pyrroles and linked by unsaturated hydrocarbon bridges often acting as multidentate ligands centered around a transition metal like Cu II, Zn II, Co II, Fe III.[1][2] Being highly conjugated molecules with many accessible energy levels, porphyrins are used in biological systems to perform light-energy conversion and modified synthetically to perform similar functions as a photoswitch or catalytic electron carriers.[1][3][4][5] Phosphorus III and V ions are much smaller than the typical metal centers and bestow distinct photochemical properties unto the porphyrin. Similar compounds with other pnictogen cores (As, Sb, Bi) or different polycyclic rings coordinated to phosphorus result in other changes to the porphyrin’s chemistry.[6][7]

Synthesis
Phosphorus core
Early experiments with group 15 elements at the porphyrin core by Barbour et al. in 1992 included syntheses of P-centered porphyrin compounds from meso-Tetra-p-tolylporphyrin (TTP). Phosphorus oxychloride (POCl3) added to H2TTP forms the P-centered porphyrin molecule [P(TTP)Cl2]+Cl-. Several chemical variants were synthesized by refluxing in solutions of pyridine solvent and alcohols to produce [P(TTP)OCH2CH3]+Cl-, [P(TTP)(O-p-C6H4OH)2]+OH-, and other similar compounds.[6] Other researchers including Poddutoori in 2015 and 2022 have used such synthetic methods to yield hypervalent phosphorus (V) bonded to porphyrin as well as axial alcohols substituents.[4][8]
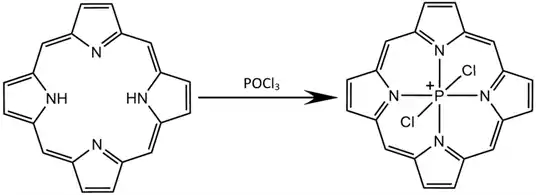
Later syntheses have been performed with other phosphorus precursors, including PhPCl2 and octaethylporphyrin (OEP) in DCM to yield [POEP(Cl)2]+Cl-.[9] PCl3/POCl3 and KPF6 yield similar porphyrin products with a PF6- counterion.[7][8] Other halogens like bromine have been used successfully in place of chlorine for this synthesis method of P-centered rings.[10] More syntheses with complex alcohols have been reported. Porphyrins functionalized with axial carbazolylvinylnaphthalimides are synthesized using similar methods to the previously described process for binding alcohols to phosphorus cores.[11] In a similar synthetic process, Susumu et al. linked several modified porphyrins in a center-to-edge bonding scheme. The chlorines on a P-centered porphyrin are first substituted by an external hydroxyphenyl group on another porphyrin. The substituent porphyrins are then refluxed with POCl3 to synthesize the final center-to-edge porphyrin array. The resulting P-centered complex consists of three porphyrins with phosphorus atoms bound at each core.[12]
Phosphaporphyrins
Synthesis of a phosphole-substituted porphyrin or phosphaporphyrin involves a more complex chemical route. Phosphaporphyrins are not created using an unmodified porphyrin ring as a synthetic reagent. As reported by Matano and Imahori in 2008, a phosphaporphyrin is constructed with a phosphole linked to two pyrrole functional groups which is then bound to another pyrrole molecule. Specifically, addition of 2,5-bis(hydroxymethyl)-1-phenyl-1-thiophosphole to excess pyrrole in the presence of BF3·OEt2 results in the phosphatripyrrane precursor. A phosphaporphyrinogen ring is formed through dehydration condensation of the phosphatripyrrane with a 2,5-difunctionalized pyrrole. Dichloro-dicyanobenzoquinone (DDQ) oxidation of the product yields the highly conjugated 18π-system phosphaporphyrin product.[13][14][15]

Metals can be coordinated in the core of the phosphaporphyrin by introducing metal salts.[15] Rhodium metal is very easily inserted into the core of a phosphaporphyrin without the presence of a stabilizing thiophene-substituted pyrrole.[16]
Properties
Several varieties of the P-centered porphyrin exist. The porphyrin with a core phosphorus (V) ion can be tuned with additional substituents added to either the outside of the polycyclic ring system or axially to the core phosphorus. Meso-substituted porphyrins like meso-tetra-p-tolylporphyrin (TTP) and octaethylporphyrin (OEP) are often used in synthesis of the core phosphorus porphyrin.[6][9] Substituents on the hypervalent phosphorus also result in the existence of a diverse array of molecules with varying properties. Axial substituents on the phosphorus include a wide variety of alkyl (-CH3, -CH2CH3), alkoxy (-OCH3, -OCH2CH3), aryl (-C6H5), and halide functional groups. Crystallographic experiments reveal that substituents affect the structure properties of these complexes. The porphyrin P-N bond distances decrease as the electronegativity of the axial substituents increase.[7] Porphyrins bound to unnaturally small ions at the core result in ruffling, a deviation in position of the carbon atoms from the median plane of the aromatic conjugated system. This phenomenon, like saddling and doming, has been observed as well with small transition metal ions like nickel II.[17] The ruffling effect of phosphorus (V) in a porphyrin is apparent because of the small size of the ion. More electronegative axial groups result in greater ruffling while large, sterically bulky groups have a similar effect. The degree to which various substituents cause ruffling was determined by Akiba et al. in 2001.[7]
Phosphaporphyrins possess phospholes usually bound to a phenyl group resting above the porphyrin plane in a trigonal pyramidal molecular geometry as is typical of phosphorus centers. In addition to the bound phenyl group, these molecules may also possess a metal ion core that coordinates to the three pyrroles and phosphole and distorts the naturally planar molecule.[16] Very negative nucleus independent chemical shift (NCIS) values used to quantify aromaticity indicate the aromatic character of the phosphaporphyrins. These values however are more positive than NICS values for the undistorted four-pyrrole structure, which is a result of the less planar π-system in phosphaporphyrins.[16]
Photoelectrochemical properties
Unmodified porphyrins have been identified and used in spectroscopic studies because of their highly conjugated π-systems. Isolated monolayers of porphyrins with dicationic transition metals yield similar spectroscopic data to those suspended in dilute solution.[1] Axial ligand substitution of the metal was proposed as a method of varying the electrochemical properties and has since been done extensively with phosphorus ions.[4]
Phosphorus core porphyrins have been researched extensively to assess their photo and electrochemical properties. Axial groups bonded to the core phosphorus exert influence on the oxidation and reduction potentials of porphyrins. Like the electronegativity effects of the axial group on P-N bonding distance and plane ruffling, electronegativity effects the molecular redox potentials. Akiba et al. in 2002 were first to quantify the redox potentials of various porphyrin molecules with different axial groups. Generally, as the electronegativity of the groups increases, the oxidative and reductive potentials become more positive indicating the complex’s ability to accept an electron more easily.[7] A 2022 study by Sharma et al. compounded the axial group electrochemical effects with the effects of outer porphyrin ring aryl substituents to determine their overall influence on P-centered porphyrin electrochemistry. More electron-withdrawing groups on the outside of the porphyrin like 3,4,5-trimethoxyphenyl resulted in a greater red-shift in the absorption spectra as opposed to less withdrawing groups like a simple phenyl. Additionally, the high oxidation potential of the modified porphyrins allows for their use in electrochemical applications. Artificial photosynthetic systems can be fine-tuned by the addition of different functional groups for catalysis and energy storage.[8][18] Poddutoori et al. in 2015 explored the electrochemical applications of the p-core porphyrins deposited with Ir(III)Cp on tin (II) oxide to form a pre-catalyst for water-splitting reactions. The ability to form an effective catalytic system is a result of the high redox potential (1.62-1.65V) of the phosphorus-modified porphyrin bound to tin (II) oxide.[4] In 2016, he devised a similar application for p-centered porphyrins with tin (IV) oxide and Mn(II)typ. Photooxidation of the Mn complex allows the reduction of the tin(IV) ion to tin(III) through the porphyrin as an electron carrier in a similar fashion to the 2015 publication.[5] The photoreduction applications mimic those of the natural porphyrin role in photosynthesis; however, the phosphorus (V) allows for tuning and more wide-ranging applications than transition metal ions.[1][4][5]
Phosphaporphyrins have been studied to a lesser degree. UV-vis and cyclic voltammetry (CV) experiments with phosphaporphyrins reveal that the single pyrrole to phosphole substitution narrows the HOMO and LUMO gap in an 18 π-system, allowing for a more easily accessible excited state and unique electrochemical applications.[13][14] The phosphole component has been shown by Reau et al. in 2002 to be prone to hyperpolarizability. This characteristic is more true of phospholes when bound to a palladium II ion as is often the case in porphyrins. Nonlinear optical (NLO) properties like molecular hyperpolarizability in metal-phosphole hybrids yield unique electrochemical applications.[14][19] A variety of phosphole-metal complexes have been synthesized to bridge copper II and silver I metal ions through phosphorus-containing chelating ligands.[20][21] The new electrochemically stabile complexes indicate the importance of the NLO properties that are unique to phosphaporphyrins by virtue of their phosphorus-integrated π-system.[14]
Photochemical applications
As early as 2002, axial substituents on the phosphorus core have been utilized for photochemistry.[3][18] Reddy and Maiya pioneered the use of a p-centered porphyrin with azobenzene substituents capable of reversible (E/Z) isomerization upon irradiation at a wavelength of 345 nm. The process of isomerization is made possible through quenching of the fluorescence by the highly conjugated porphyrin ring and the reversibility of the isomerization. The high yield of both isomers after several iterations of the reaction indicates that the photoswitching is a reliable, stable process.[8]

Photovoltaic devices using NiO have also been made more efficient by hole injection of phosphorus porphyrins, yielding ground state reactants that are regenerated at a much faster rate than is typical for a TiO2 cell.[22]
Biological applications
Photodynamic therapy (PDT) is used to target and destroy DNA in cases of extreme cell proliferation as is common in cancer. Porphyrins are used to generate radical oxygen species in the body to degrade nucleotides, stopping proliferation.[23][24] Tetraphenylporphyrin phosphorus (V) complexes with DNA and possesses an oxidation potential larger than guanine(1.4-1.8V > 1.24V). Upon coupling to DNA fragments through electrostatic interaction, a p-centered porphyrin electron is excited with 365 nm radiation to the S2 state and proceeds down an energy cascade via two mechanisms to degrade guanine. The first mechanism involves electron transfer that degrades a sequence of consecutive guanine nucleotides while the second mechanism proceeds via oxygen radical formation that indiscriminately destroys guanine residues in DNA.[23]

Reactivity
P-centered porphyrins typically react in redox reactions where they serve as tunable intermediate electron carriers in biological reactions for DNA degradation as well as other industrial catalytic reactions.[23] Tn (II/IV) oxide is photoreduced by iridium and manganese complexes through p-centered porphyrin electron carriers in catalysis.[4][5] Photovoltaic cells involving NiO complexes supplemented with high oxidation potential p-centered porphyrins improve cell efficiency as is the case in indium tin oxide cells with porphyrins containing axial carbazolylvinylnaphthalimides bound to a core phosphorus.[11][22]
Phosphorus (V) porphyrins are particularly good electron carriers in redox reactions because of their adjustable reduction potential. Poddutoori et al. in 2021 investigated the electron transfer of naphthalene, a commonly excited molecule in photochemistry, to octaethylporphyrin and TEP. The relatively low potentials of the porphyrins yield highly energetic charge-separated states upon the transfer of the electron from naphthalene. The axial and peripheral substituent diversity is key to accessing the wide range of charge-separated states and electron transfer across electronically diverse reactants to form a great variety of redox products.[9]

Phosphaporphyrins after synthesis can be complexed with metals like the unmodified porphyrin molecule. Nickel, palladium, and platinum can be coordinated as the metal center of a phosphaporphyrin by reacting the conjugated ring with metal salts like Ni(cod)2, Pd(dba)2, and Pt(dba)2 in DCM/dichlorobenzene respectively.[15]
Derivatives and structural analogs
Other pnictogen ion cores
Phosphorus (V) is a prevalent ion center in modified porphyrin complexes but is not the only group 15 element that has been used in place of a transition metal ion. Antimony and bismuth have also been identified as suitable porphyrin cores as early as 1991 by Barbour et al.[6] Stronger electron donating from the less electronegative antimony results in a more positive reduction potential than in the phosphorus ion. Sterically speaking, arsenic (V), being a larger ion than phosphorus (V), possesses a more stable, planar coordinated porphyrin as opposed to the ruffled porphyrin coordinated to phosphorus. Oppositely, a pnictogen element too large to neatly coordinate into the porphyrin hole like bismuth causes a disruption of symmetry in the ring.[6][7]

Calixphyrins
Calixphyrins are analogous to porphyrins with two of the hydrocarbon bridges between pyrroles fully saturated. Like phosphaporphyrin, a calixphyrin pyrrole can be substituted with a phosphole to form a P-centered calixphyrin. The results of increased saturation are mixed sp2 and sp3 hybridization of the ring carbons and the extension of the metal-nitrogen bond length.[15][25]
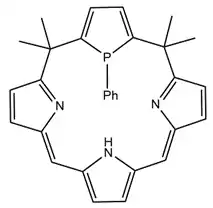
Matano et al. in 2006 used the first ever P,S-hybridized calixphyrin coordinated to palladium to catalyze the Heck reaction to form substituted alkenes.[26] Like porphyrins, electronic properties of this complex are also tunable through the influence of various functional groups.[27] In 2009, these molecules were examined in comparison to porphyrins due to their more restricted π-systems and carefully characterized with a focus on the tunability of their electronic properties with different metals and substituents.[15][27]
Calixpyrroles
Calixpyrroles have all four hydrocarbon bridges fully saturated, breaking the conjugation between heterocycles present in other porphyrinoids. A phosphole replacing a pyrrole allows for similar chemistry to other phophsaporphyrinoids with increased flexibility of the tetradentate ligand by virtue of sp3 hybridization.[15]

Isophlorins
Isophlorins are less stable nonaromatic complexes that engage in similar chemistry to the porphyrinoids. The 20π systems are synthesized from 18π porphyrins via redox-coupled complexation. Although the 4πn conjugated system would suggest that the molecule exhibits antiaromatic character, geometric and magnetic criteria confirm that the complex is nonaromatic.[28]

References
- 1 2 3 4 Schick, Alan; Schreiman, Irwin C.; Wagner, Richard W.; Lindsey, Jonathan S.; and Bocian, David F. (1989). “Spectroscopic characterization of porphyrin monolayer assemblies”. Journal of the American Chemical Society 111 (4): 1344-1350. DOI: 10.1021/ja00186a030
- ↑ Gong, Xianchang; Milic, Tatjana; Xu, Chang; Batteas, James D.; and Drain, Charles Michael. (2002). “Preparation and Characterization of Porphyrin Nanoparticles”. Journal of the American Chemical Society 124 (48): 14290-14291. DOI: 10.1021/ja027405z
- 1 2 Reddya, D. Raghunath and Maiya, Bhaskar G. (2000). “A molecular photoswitch based on an ‘axial-bonding’ type phosphorus(v) porphyrin”. Chemical Communications 117-118 DOI: 10.1039/b007784o
- 1 2 3 4 5 6 Poddutoori, Prashanth K.; Thomsen, Julianne M.; Milot, Rebecca L.; Sheehan, Stafford W.; et al. (2015). “Interfacial electron transfer in photoanodes based on phosphorus(v) porphyrin sensitizers co-deposited on SnO2 with the Ir(III)Cp* water oxidation precatalyst”. Journal of Material Chemistry 3: 3868-3879. DOI: DOI: 10.1039/c4ta07018f
- 1 2 3 4 Poddutoori, Prashanth K.; Lim, Gary N.; Pilkington, Melanie; D’Souza, Francis; and van der Est, Art. (2016). “Phosphorus(V) Porphyrin-Manganese(II) Terpyridine Conjugates: Synthesis, Spectroscopy, and Photo-Oxidation Studies on a SnO2 Surface”. Inorganic Chemistry 55 (21): 11383-11395. DOI: 10.1021/acs.inorgchem.6b01924
- 1 2 3 4 5 Barbour, Tanya; Belcher, Warwick J.; Brothers, Penelope J.; Rickard, Clifton E. F.; and Ware, David C. (1992). “Preparation of Group 15 (Phosphorus, Antimony, and Bismuth) Complexes of meso-Tetra-p-tolylporphyrin (TTP) and X-ray Crystal Structure of [Sb(TTP)(OCH(CH3)2)2]Cl”. Inorganic Chemistry 31 (5): 746-754. DOI: 10.1021/ic00031a011
- 1 2 3 4 5 6 Akiba, Kin-ya; Nadano, Ryo; Satoh, Wataru; Yamamoto, Yohsuke; Nagase, Shigeru; Ou, Zhongping; Tan, Xiaoyu; and Kadish, Karl M. (2001). “Synthesis, Structure, Electrochemistry, and Spectroelectrochemistry of Hypervalent Phosphorus(V) Octaethylporphyrins and Theoretical Analysis of the Nature of the PO Bond in P(OEP)(CH2CH3)(O)”. Inorganic Chemistry 40 (22): 5553-5567. DOI: 10.1021/ic010595e
- 1 2 3 4 Sharma, Jatan K.; Bayard, Brandon J.; Zosel, Nick; Ali, Syeda S.; Holzer, Noah; Nesterov, Vladimir N.; Karr, Paul A.; D’Souza, Francis; and Poddutoori, Prashanth K. (2022). “Hypervalent Phosphorus(V) Porphyrins with meso-Methoxyphenyl Substituents: Significance of the Number and Position of Methoxy Groups in Promoting Intramolecular Charge Transfer”. Inorganic Chemistry 61 (42): 16573–16585. DOI: 10.1021/acs.inorgchem.2c01648
- 1 2 3 Poddutoori, Prashanth K; Bayard, Brandon J.; Holzer, Noah; Seetharaman, Sairaman; Zarrabi, Niloofar; Weidner, Nathan; Karr, Paul A; and D’Souza, Francis. (2021). “Rational Design and Synthesis of OEP and TPP Centered Phosphorus(V) Porphyrin–Naphthalene Conjugates: Triplet Formation via Rapid Charge Recombination”. Inorganic Chemistry 60 (23): 17952-17965. DOI: 10.1021/acs.inorgchem.1c02531
- ↑ Furuyama, Taniyuki; Maeda, Kazuya; Maeda, Hajime; and Segi, Masahito. (2019). “Chemoselective Synthesis of Aryloxy-Substituted Phthalocyanines”. The Journal of Organic Chemistry 84 (21): 14306-14312. DOI: 10.1021/acs.joc.9b02126
- 1 2 Zhan, Yong; Cao, Kaiyu; Wang, Chenguang; Jia, Junhui; et al. (2012). “Synthesis and photophysical properties of phosphorus(v) porphyrins functionalized with axial carbazolylvinylnaphthalimides”. Organic Biomolecular Chemistry 10, 8701-8709. DOI: 10.1039/c2ob26478a
- ↑ Susumu, Kimihiro; Tanaka, Kazuyoshi; Shimidzu, Takeo; Takeuchi, Yasuko; and Segawa, Hiroshi. (1999). “Synthesis and photophysical properties of “center-to-edge” type phosphorus(V) porphyrin arrays”. J. Chem. Soc., Perkin Trans. 2: 1521–1529. DOI: 10.1039/A809840I
- 1 2 Matano, Yoshihiro; Nakabuchi, Takashi; Miyajima, Tooru; Imahori, Hiroshi; and Nakano, Haruyuki. (2006). “Synthesis of a Phosphorus-Containing Hybrid Porphyrin”. Organic Letters 8 (25): 5713-5716. DOI: 10.1021/ol0622763
- 1 2 3 4 Matano, Yoshihiro and Imahori, Hiroshi. (2009). “Design and synthesis of phosphole-based π systems for novel organic materials”. Organic Biomolecular Chemistry 7: 1258-1271. DOI: 10.1039/b819255n
- 1 2 3 4 5 6 Matano, Yoshihiro and Imahori, Hiroshi. (2009). “Phosphole-containing calixpyrroles, calixphyrins, and porphyrins: synthesis and coordination chemistry”. Accounts of Chemical Research 42 (8): 1193-1204. DOI: 10.1021/ar900075e
- 1 2 3 Matano, Yoshihiro; Nakabuchi, Takashi; and Imahori, Hiroshi. (2010). “Synthesis, structures, and aromaticity of phosphole-containing porphyrins and their metal complexes”. Pure Appl. Chem. 82: 583-593. DOI: 10.1351/PAC-CON-09-08-05
- ↑ Conradie, Jeanet and Ghosh, Abhik. (2017). “Energetics of Saddling versus Ruffling in Metalloporphyrins: Unusual Ruffled Dodecasubstituted Porphyrins”. ACS Omega 2 (10): 6708-6714. DOI: 10.1021/acsomega.7b01004
- 1 2 Meshkov, Ivan N.; Bulach, Véronique; Gorbunova, Yulia G.; Gostev, Fedor E.; Nadtochenko, Victor A.; Tsivadze, Aslan Yu; and Hosseini, Mir Wais. (2017). “Tuning photochemical properties of phosphorus(v) porphyrin photosensitizers”. Chemical Communications 53: 9918-9921. DOI: 10.1039/c7cc06052a
- ↑ Fave, Claire; Hissler, Muriel; Sénéchal, Katell; Ledoux, Isabelle; Zyssb, Joseph; and Réau, Régis. (2002). “Ligandtrans-effect: using an old concept as a novel approach to bis(dipolar) NLO-phores”. Chemical Communications 16: 1674-1675. DOI: 10.1039/B203149C
- ↑ Nohra, Brigitte; Rodriguez-Sanz, Elena; Lescop Dr., Christophe; and Réau, Régis. (2008). “Chemistry of Bridging Phosphanes: CuI Dimers Bearing 2,5-Bis(2-pyridyl)phosphole Ligands”. Chemistry Europe 14 (11): 3391-3403. DOI: 10.1002/chem.200701423
- ↑ Welsch, Stefan; Lescop, Christophe; Scheer, Manfred; and Réau, Régis. (2008). “AgI Bimetallic Molecular Clips with Adaptive Coordination Behavior for Supramolecular Chemistry”. Inorganic Chemistry 47 (19): 8592-8594. DOI: 10.1021/ic801222j
- 1 2 Borgström, Magnus; Blart, Errol; Boschloo, Gerrit; Mukhtar, Emad; Hagfeldt, Anders; Hammarström, Leif; and Odobel, Fabrice. (2005). “Sensitized Hole Injection of Phosphorus Porphyrin into NiO: Toward New Photovoltaic Devices”. The Journal of Physical Chemistry B 109 (48): 22928-22934. DOI: 10.1021/jp054034a
- 1 2 3 Hirakawa, F. Kazutaka; Kawanishi, Shosuke; Hirano, Toru; Segawa, Hiroshi. (2007). “Guanine-specific DNA oxidation photosensitized by the tetraphenylporphyrin phosphorus(V) complex via singlet oxygen generation and electron transfer”. The Journal of Photochemistry and Photobiology B: Biology 87 (3): 209-217. DOI: 10.1016/j.jphotobiol.2007.04.001
- ↑ Besaratinia, Ahmad; Bates, Steven E.; Synold, Timothy W.; and Pfeifer, Gerd P. (2004). “Similar Mutagenicity of Photoactivated Porphyrins and Ultraviolet A Radiation in Mouse Embryonic Fibroblasts: Involvement of Oxidative DNA Lesions in Mutagenesis”. Biochemistry 43 (49): 15557-15566. DOI: 10.1021/bi048717c
- ↑ Matano, Yoshihiro; Miyajima, Tooru; Ochi, Noriaki; Nakabuchi, Takashi; Shiro, Motoo; Nakao, Yoshihide; Sakaki, Shigeyoshi; and Imahori, Hiroshi. (2008). “Syntheses, Structures, and Coordination Chemistry of Phosphole-Containing Hybrid Calixphyrins: Promising Macrocyclic P,N2,X-Mixed Donor Ligands for Designing Reactive Transition Metal Complexes”. Journal of the American Chemical Society 130 (3): 990-1002. DOI: 10.1021/ja076709o
- ↑ Matano, Yoshihiro; Miyajima, Tooru; Nakabuchi, Takashi; Imahori, Hiroshi; Ochi, Noriaki; and Sakaki, Shigeyoshi. (2006). “Phosphorus-Containing Hybrid Calixphyrins: Promising Mixed-Donor Ligands for Visible and Efficient Palladium Catalysts”. Journal of the American Chemical Society 128 (36): 11760-11761. DOI: 10.1021/ja0640039
- 1 2 Matano, Yoshihiro; Fujita, Masato; Miyajima, Tooru; and Imahori, Hiroshi. (2009). “Meso-Substituent Effects on Redox Properties of the 5,10-Porphodimethene-Type P,S,N2-Hybrid Calixphyrins and Their Metal Complexes”. Organometallics 28 (21): 6213-6217. DOI: 10.1021/om900745t
- ↑ Matano, Yoshihiro; Nakabuchi, Takashi; Fujishige, Shinya; Nakano, Haruyuki; and Imahori, Hiroshi. (2008). “Redox-Coupled Complexation of 23-Phospha-21-thiaporphyrin with Group 10 Metals: A Convenient Access to Stable Core-Modified Isophlorin−Metal Complexes”. Journal of the American Chemical Society 130 (49): 16446-16447. DOI: 10.1021/ja807742g