
Protein crystallization is the process of formation of a regular array of individual protein molecules stabilized by crystal contacts. If the crystal is sufficiently ordered, it will diffract. Some proteins naturally form crystalline arrays, like aquaporin in the lens of the eye.[1][2]
In the process of protein crystallization, proteins are dissolved in an aqueous environment and sample solution until they reach the supersaturated state.[3] Different methods are used to reach that state such as vapor diffusion, microbatch, microdialysis, and free-interface diffusion. Developing protein crystals is a difficult process influenced by many factors, including pH, temperature, ionic strength in the crystallization solution, and even gravity.[3] Once formed, these crystals can be used in structural biology to study the molecular structure of the protein, particularly for various industrial or medical purposes.[4][5]
Development of protein crystallization
For over 150 years, scientists from all around the world have known about the crystallization of protein molecules.[6]
In 1840, Friedrich Ludwig Hünefeld accidentally discovered the formation of crystalline material in samples of earthworm blood held under two glass slides and occasionally observed small plate-like crystals in desiccated swine or human blood samples. These crystals were named as 'haemoglobin', by Felix Hoppe-Seyler in 1864. The seminal findings of Hünefeld inspired many scientists in the future.[7]
In 1851, Otto Funke described the process of producing human haemoglobin crystals by diluting red blood cells with solvents, such as pure water, alcohol or ether, followed by slow evaporation of the solvent from the protein solution. In 1871, William T. Preyer, Professor at University of Jena, published a book entitled Die Blutkrystalle (The Crystals of Blood), reviewing the features of haemoglobin crystals from around 50 species of mammals, birds, reptiles and fishes.[7]
In 1909, the physiologist Edward T. Reichert, together with the mineralogist Amos P. Brown, published a treatise on the preparation, physiology and geometrical characterization of haemoglobin crystals from several hundreds animals, including extinct species such as the Tasmanian wolf.[7] Increasing protein crystals were found.
In 1934, John Desmond Bernal and his student Dorothy Hodgkin discovered that protein crystals surrounded by their mother liquor gave better diffraction patterns than dried crystals. Using pepsin, they were the first to discern the diffraction pattern of a wet, globular protein. Prior to Bernal and Hodgkin, protein crystallography had only been performed in dry conditions with inconsistent and unreliable results. This is the first X‐ray diffraction pattern of a protein crystal.[8]
In 1958, the structure of myoglobin (a red protein containing heme), determined by X-ray crystallography, was first reported by John Kendrew.[9] Kendrew shared the 1962 Nobel Prize in Chemistry with Max Perutz for this discovery.[4]
Now, based on the protein crystals, the structures of them play a significant role in biochemistry and translational medicine.
The basics of protein crystallization

The theory of protein crystallization
Protein crystallization is governed by the same physics that governs the formation of inorganic crystals. For crystallization to occur spontaneously, the crystal state must be favored thermodynamically. This is described by Gibb's free energy (∆G), defined as ∆G = ∆H- T∆S, which captures how the energetics of a process, ∆H, trades off with the corresponding change in entropy, ∆S.[10] Entropy, roughly, describes the disorder of a system. Highly ordered states, such as protein crystals, are disfavored thermodynamically compared to more disordered states, such as solutions of proteins in solvent, because the transition to a more ordered state would decrease the total entropy of the system (positive ∆S). For crystals to form spontaneously, the ∆G of crystal formation must be negative. In other words, the entropic penalty must be paid by a corresponding decrease in the total energy of the system (∆H). Familiar inorganic crystals such as sodium chloride spontaneously form at ambient conditions because the crystal state decreases the total energy of the system. However, crystallization of some proteins under ambient conditions would both decrease the entropy (positive ∆S) and increase the total energy (positive ∆H) of the system, and thus does not occur spontaneously. To achieve crystallization of such proteins conditions are modified to make crystal formation energetically favorable. This is often accomplished by creation of a supersaturated solution of the sample.[3]
A molecular view going from solution to crystal
Crystal formation requires two steps: nucleation and growth.[3] Nucleation is the initiation step for crystallization.[3] At the nucleation phase, protein molecules in solution come together as aggregates to form a stable solid nucleus.[3] As the nucleus forms, the crystal grows bigger and bigger by molecules attaching to this stable nucleus.[3] The nucleation step is critical for crystal formation since it is the first-order phase transition of samples moving from having a high degree of freedom to obtaining an ordered state (aqueous to solid).[3] For the nucleation step to succeed, the manipulation of crystallization parameters is essential. The approach behind getting a protein to crystallize is to yield a lower solubility of the targeted protein in solution.[3] Once the solubility limit is exceeded and crystals are present, crystallization is accomplished.[3]
Methods of protein crystallization
Vapor diffusion
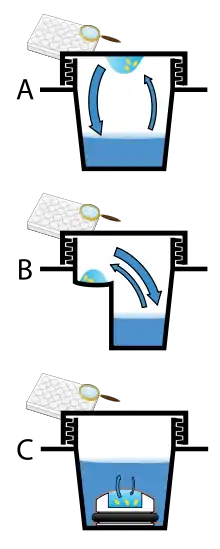
Vapor diffusion is the most commonly employed method of protein crystallization. In this method, droplets containing purified protein, buffer, and precipitant are allowed to equilibrate with a larger reservoir containing similar buffers and precipitants in higher concentrations. Initially, the droplet of protein solution contains comparatively low precipitant and protein concentrations, but as the drop and reservoir equilibrate, the precipitant and protein concentrations increase in the drop. If the appropriate crystallization solutions are used for a given protein, crystal growth occurs in the drop.[11][12] This method is used because it allows for gentle and gradual changes in concentration of protein and precipitant concentration, which aid in the growth of large and well-ordered crystals.
Vapor diffusion can be performed in either hanging-drop or sitting-drop format. Hanging-drop apparatus involve a drop of protein solution placed on an inverted cover slip, which is then suspended above the reservoir. Sitting-drop crystallization apparatus place the drop on a pedestal that is separated from the reservoir. Both of these methods require sealing of the environment so that equilibration between the drop and reservoir can occur.[11][13]
Microbatch
A microbatch usually involves immersing a very small volume of protein droplets in oil (as little as 1 µl). The reason that oil is required is because such low volume of protein solution is used and therefore evaporation must be inhibited to carry out the experiment aqueously. Although there are various oils that can be used, the two most common sealing agent are paraffin oils (described by Chayen et al.) and silicon oils (described by D’Arcy). There are also other methods for microbatching that don't use a liquid sealing agent and instead require a scientist to quickly place a film or some tape on a welled plate after placing the drop in the well.
Besides the very limited amounts of sample needed, this method also has as a further advantage that the samples are protected from airborne contamination, as they are never exposed to the air during the experiment.
Microdialysis
Microdialysis takes advantage of a semi-permeable membrane, across which small molecules and ions can pass, while proteins and large polymers cannot cross. By establishing a gradient of solute concentration across the membrane and allowing the system to progress toward equilibrium, the system can slowly move toward supersaturation, at which point protein crystals may form.
Microdialysis can produce crystals by salting out, employing high concentrations of salt or other small membrane-permeable compounds that decrease the solubility of the protein. Very occasionally, some proteins can be crystallized by dialysis salting in, by dialyzing against pure water, removing solutes, driving self-association and crystallization.
Free-interface diffusion
This technique brings together protein and precipitation solutions without premixing them, but instead, injecting them through either sides of a channel, allowing equilibrium through diffusion. The two solutions come into contact in a reagent chamber, both at their maximum concentrations, initiating spontaneous nucleation. As the system comes into equilibrium, the level of supersaturation decreases, favouring crystal growth.[14]
Factors influencing protein crystallization
pH
The basic driving force for protein crystallization is to optimize the number of bonds one can form with another protein through intermolecular interactions.[3] These interactions depend on electron densities of molecules and the protein side chains that change as a function of pH.[10] The tertiary and quaternary structure of proteins are determined by intermolecular interactions between the amino acids’ side groups, in which the hydrophilic groups are usually facing outwards to the solution to form a hydration shell to the solvent (water).[10] As the pH changes, the charge on these polar side group also change with respect to the solution pH and the protein's pKa. Hence, the choice of pH is essential either to promote the formation of crystals where the bonding between molecules to each other is more favorable than with water molecules.[10] pH is one of the most powerful manipulations that one can assign for the optimal crystallization condition.
Temperature
Temperature is another interesting parameter to discuss since protein solubility is a function of temperature.[15] In protein crystallization, manipulation of temperature to yield successful crystals is one common strategy. Unlike pH, temperature of different components of the crystallography experiments could impact the final results such as temperature of buffer preparation,[16] temperature of the actual crystallization experiment, etc.
Chemical Additives
Chemical additives are small chemical compounds that are added to the crystallization process to increase the yield of crystals.[17] The role of small molecules in protein crystallization had not been well thought of in the early days since they were thought of as contaminants in most case.[17] Smaller molecules crystallize better than macromolecules such as proteins, therefore, the use of chemical additives had been limited prior to the study by McPherson. However, this is a powerful aspect of the experimental parameters for crystallization that is important for biochemists and crystallographers to further investigate and apply.[17]
Technologies assisting protein crystallization
High throughput crystallization screening [18]
High through-put methods exist to help streamline the large number of experiments required to explore the various conditions that are necessary for successful crystal growth. There are numerous commercial kits available for order which apply preassembled ingredients in systems guaranteed to produce successful crystallization. Using such a kit, a scientist avoids the hassle of purifying a protein and determining the appropriate crystallization conditions.
Liquid-handling robots can be used to set up and automate large number of crystallization experiments simultaneously. What would otherwise be slow and potentially error-prone process carried out by a human can be accomplished efficiently and accurately with an automated system. Robotic crystallization systems use the same components described above, but carry out each step of the procedure quickly and with a large number of replicates. Each experiment utilizes tiny amounts of solution, and the advantage of the smaller size is two-fold: the smaller sample sizes not only cut-down on expenditure of purified protein, but smaller amounts of solution lead to quicker crystallizations. Each experiment is monitored by a camera which detects crystal growth.[12]
Protein engineering
Proteins can be engineered to improve the chance of successful protein crystallization by using techniques like Surface Entropy Reduction[19] or engineering in crystal contacts.[20] Frequently, problematic cysteine residues can be replaced by alanine to avoid disulfide-mediated aggregation, and residues such as lysine, glutamate, and glutamine can be changed to alanine to reduce intrinsic protein flexibility, which can hinder crystallization..
Applications of protein crystallography
Macromolecular structures can be determined from protein crystal using a variety of methods, including X-Ray Diffraction/X-ray crystallography, Cryogenic Electron Microscopy (CryoEM) (including Electron Crystallography and Microcrystal Electron Diffraction (MicroED)), Small-angle X-ray scattering, and Neutron diffraction. See also Structural biology.
Crystallization of proteins can also be useful in the formulation of proteins for pharmaceutical purposes.[21]
See also
References
- ↑ Schey KL, Wang Z, L Wenke J, Qi Y (May 2014). "Aquaporins in the eye: expression, function, and roles in ocular disease". Biochimica et Biophysica Acta (BBA) - General Subjects. 1840 (5): 1513–1523. doi:10.1016/j.bbagen.2013.10.037. PMC 4572841. PMID 24184915.
- ↑ Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T (December 2005). "Lipid-protein interactions in double-layered two-dimensional AQP0 crystals". Nature. 438 (7068): 633–638. Bibcode:2005Natur.438..633G. doi:10.1038/nature04321. PMC 1350984. PMID 16319884.
- 1 2 3 4 5 6 7 8 9 10 11 McPherson A, Gavira JA (January 2014). "Introduction to protein crystallization". Acta Crystallographica. Section F, Structural Biology Communications. 70 (Pt 1): 2–20. doi:10.1107/s2053230x13033141. PMC 3943105. PMID 24419610.
- 1 2 Blundell TL (July 2017). "Protein crystallography and drug discovery: recollections of knowledge exchange between academia and industry". IUCrJ. 4 (Pt 4): 308–321. doi:10.1107/s2052252517009241. PMC 5571795. PMID 28875019.
- ↑ Tripathy D, Bardia A, Sellers WR (July 2017). "Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors". Clinical Cancer Research. 23 (13): 3251–3262. doi:10.1158/1078-0432.ccr-16-3157. PMC 5727901. PMID 28351928.
- ↑ McPherson A (March 1991). "A brief history of protein crystal growth". Journal of Crystal Growth. 110 (1–2): 1–10. Bibcode:1991JCrGr.110....1M. doi:10.1016/0022-0248(91)90859-4. ISSN 0022-0248.
- 1 2 3 Giegé R (December 2013). "A historical perspective on protein crystallization from 1840 to the present day". The FEBS Journal. 280 (24): 6456–6497. doi:10.1111/febs.12580. PMID 24165393.
- ↑ Tulinsky A (1996). "Chapter 35. The Protein Structure Project, 1950–1959: First Concerted Effort of a Protein Structure Determination in the U.S.". Annual Reports in Medicinal Chemistry. Elsevier. 31: 357–366. doi:10.1016/s0065-7743(08)60474-1. ISBN 9780120405312.
- ↑ Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC (March 1958). "A three-dimensional model of the myoglobin molecule obtained by x-ray analysis". Nature. 181 (4610): 662–666. Bibcode:1958Natur.181..662K. doi:10.1038/181662a0. PMID 13517261. S2CID 4162786.
- 1 2 3 4 Boyle J (January 2005). "Lehninger principles of biochemistry (4th ed.): Nelson, D., and Cox, M." Biochemistry and Molecular Biology Education. 33 (1): 74–75. doi:10.1002/bmb.2005.494033010419. ISSN 1470-8175.
- 1 2 Rhodes G (2006). Crystallography Made Crystal Clear: A Guide for Users of Macromolecular Models (Third ed.). Academic Press.
- 1 2 "The Crystal Robot". December 2000. Retrieved 2003-02-18.
- ↑ McRee D (1993). Practical Protein Crystallography. San Diego: Academic Press. pp. 1–23. ISBN 978-0-12-486052-0.
- ↑ Rupp B (20 October 2009). Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. Garland Science. p. 800. ISBN 9781134064199. Retrieved 28 December 2016.
- ↑ Pelegrine DH, Gasparetto CA (February 2005). "Whey proteins solubility as function of temperature and pH". LWT - Food Science and Technology. 38 (1): 77–80. doi:10.1016/j.lwt.2004.03.013. ISSN 0023-6438.
- ↑ Chen RQ, Lu QQ, Cheng QD, Ao LB, Zhang CY, Hou H, et al. (January 2015). "An ignored variable: solution preparation temperature in protein crystallization". Scientific Reports. 5 (1): 7797. Bibcode:2015NatSR...5E7797C. doi:10.1038/srep07797. PMC 4297974. PMID 25597864.
- 1 2 3 McPherson A, Cudney B (December 2006). "Searching for silver bullets: an alternative strategy for crystallizing macromolecules". Journal of Structural Biology. 156 (3): 387–406. doi:10.1016/j.jsb.2006.09.006. PMID 17101277. S2CID 10944540.
- ↑ Lin Y (August 2018). "What's happened over the last five years with high-throughput protein crystallization screening?". Expert Opinion on Drug Discovery. 13 (8): 691–695. doi:10.1080/17460441.2018.1465924. PMID 29676184.
- ↑ Cooper DR, Boczek T, Grelewska K, Pinkowska M, Sikorska M, Zawadzki M, Derewenda Z (May 2007). "Protein crystallization by surface entropy reduction: optimization of the SER strategy". Acta Crystallographica. Section D, Biological Crystallography. 63 (Pt 5): 636–645. doi:10.1107/S0907444907010931. PMID 17452789.
- ↑ Gonen S, DiMaio F, Gonen T, Baker D (June 2015). "Design of ordered two-dimensional arrays mediated by noncovalent protein-protein interfaces". Science. 348 (6241): 1365–1368. Bibcode:2015Sci...348.1365G. doi:10.1126/science.aaa9897. PMID 26089516.
- ↑ Jen A, Merkle HP (November 2001). "Diamonds in the rough: protein crystals from a formulation perspective". Pharmaceutical Research. 18 (11): 1483–8. doi:10.1023/a:1013057825942. PMID 11758753. S2CID 21801946.
Further reading
- Cudney R (1999). "Protein Crystallization and Dumb Luck" (PDF). The Rigaku Journal. 16 (1): 1–7. Archived from the original (PDF) on 4 March 2016.
- Owens R. "Protein Crystals". Backstage Science. Brady Haran.
External links
- This page was reproduced (with modifications) with expressed consent from Dr. A. Malcolm Campbell. As of 2010, the original page can be found at Campbell AM (2003). "Protein Crystallization". Davidson, NC: Department of Biology, Davidson College.