| Soluble NSF Attachment Protein | |||||||||
|---|---|---|---|---|---|---|---|---|---|
Sec17, a yeast homolog of the human SNAP, patriciates in membrane fusion of vesicles complexes with SNARE and NSF to mediate assembly and disassembly. | |||||||||
| Identifiers | |||||||||
| Symbol | SNAP | ||||||||
| Pfam | PF14938 | ||||||||
| PROSITE | PDOC50892 PDOC50192, PDOC50892 | ||||||||
| CDD | cd15832 | ||||||||
| |||||||||
Soluble N-ethylmaleimide-Sensitive Factor Attachment Proteins (SNAP, or Sec17p in yeast) are a family of cytosolic adaptor proteins involved in vesicular fusion at membranes during intracellular transport and exocytosis. SNAPs interact with proteins of the SNARE complex and NSF to play a key role in recycling the components of the fusion complex. SNAPs are involved in the priming of the vesicle fusion complex during assembly, as well as in the disassembly following a vesicle fusion event. Following membrane fusion, the tethering SNARE proteins complex disassembles in response to steric changes originating from the ATPase NSF. The energy provided by NSF is transferred throughout the SNARE complex and SNAP, allowing the proteins to untangle, and recycled for future fusion events. Mammals have three SNAP genes: α-SNAP, β-SNAP, and γ-SNAP. α- and γ-SNAP are expressed throughout the body, while β-SNAP is specific to the brain. The yeast homolog of the human SNAP is Sec17, the structural diagram of which is included on this page.
Function
The function of SNAP proteins have been primarily related to the role which the play in the assemble and disassembly of SNARE complex required for vesicle fusion events. According to the SNARE hypothesis developed in the early 1990s, SNAP protein are localized to the membranes and are central in mediating Ca2+ dependent vesicle fusion at these sites. SNAPs associate with the proteins of the SNARE (SNAP REceptor) complex, a class of type II integral membrane protein, as well as the ATPase NSF, largely based on electrostatic interactions.[1][2] The interaction of the SNAPs with SNAREs takes place before interaction of the complex with NSF (Sec18 in yeast) suggesting a sequence for the priming assembly may be necessary.[1][3] The assembled complex which includes SNAP, SNARE, and NSF is known as the 20S complex. Some of the first proteins identified as the receptors of SNAPs were syntaxin 1, SNAP-25 (synaptosome associated protein, 25kDa), and VAMP (synaptobrevin).[4] These proteins contain transmembrane regions that can be found in both intracellular vesicles and as part of extracellular trafficking machinery. Figure 1 shows interactions of the vesicular and membrane SNARE proteins with NSF and SNAP in the assembly, fusion, and disassembly process that accompanies vesicle fusion events.
Initial binding of NSF to SNAP been is likely related to interactions of the 63 N-terminal and 37 C-terminal amino acid residues of SNAP with NSF protein.[5] The interaction with SNAP stimulates the ATPase activity of the NSF when assembled into the 20S complex, and ultimately leads to ATP hydrolysis that result in the disruption of the heterooligomeric complex.[6] This has the potential to reduce or block synaptic transmission, ultimately leading to the loss of signaling downstream. Further information on this is included in the toxicology section below.
While assembly of the complex can take place under only conditions where a components and a membrane is present, disassembly requires that NSF can hydrolyze ATP.[1] Use of chelating agents, non-hydrolysable analogues of GTP, or application of an alkylating agent N-ethylmaleimide (NEM), therefore, has been used to demonstrate prevention of vesicle fusion in vitro.[4] Blocking the assembly of the 20S complex also prevents the ATP-hydrolysis reaction from taking place at NSF.
Limitations of the Original SNARE Theory of Vesicle Fusion

The SNARE theory of vesicle fusion, describes the action mechanism of SNAREs, SNAP, and NSF, but does not completely explain all known vesicle fusion related kinetics. The theory was first put forth by James Rothman and co-workers starting in the early 1990s and predicted that SNAPs and NSF recognized paired vesicle-SNARE (v-SNARE)/ target-SNARE (t-SNARE) complexes at membranes and bound to them thus creating the 20S complex. These complex form similar structures for both synaptic and vacuolar systems including the Golgi transport.
Data generated experimentally in recent years lead some to question the completeness of the model. Although it was known since the 1960s that Ca2+ influx was responsible for synaptic signaling, a collaboration in 1992 between Thomas Südhof and Reinhardt Jahn tied the link between calcium, SNARE complexes and synaptic signaling, suggesting that vesicle fusion events were not rate limited by the SNARE complex formation as previously thought. At the time, the SNARE complex model could not account for the rapid release of neurotransmitters into synaptic clefts, as the complex disassociation and recycling was thought to be rate limiting for further vesicle fusion.
Further studies demonstrated that the ATP hydrolysis step occurs prior to a calcium ion mediated fusion event, and thus revealing, that SNAP and NSF proteins initiate disassembly the 20S complex before the docking event takes place directly at the membrane. The existence of these ATP primed vesicles for fusion at the pre-synaptic membrane is facilitated by the interactions of SNAP and NSF.
It is now understood that the 20S complex does not disassociate immediately following ATP hydrolysis, but rather remains tethered until intracellular Ca2+ achieve significantly high levels to facilitate docking. A depolarizing current that leads to the opening of voltage dependent ion channels permits the influx of Ca2+ into the cell where the molecular clamp protein (a SNARE) called synaptotagmin acts in a Ca2+ sensitive manner to facilitate fusion of the vesicle to the membrane up to a rate of one vesicle per 100us. The exocytosis of neurotransmitters as regulated by Ca2+ therefore, has faster kinetics than would be possible by the SNARE-recycling model alone. Figure 1 summarizes the updated model of the SNARE hypothesis.
Significance in Toxicology
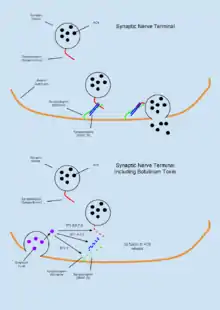
The 20S complex is a known target for Clostridium neurotoxins including Botulinum A, C. and E, which block synaptic transmission by disrupting the complex and preventing neurotransmitter release into the synaptic space. The disruption to synaptic transmission is caused by serotype B toxins cleaving VAMP-2/synaptobrevin-2, but not type 1 SNARE proteins. Botulinum toxins do not directly interact with SNAP, but the indirectly impact its ability to assemble into the 20S complex leading to impaired synaptic transmission at the neuromuscular junction. The blocking of acetylcholine release onto the endplate leads to muscle paralysis and, if left untreated, death. Poisoning by botulinum toxin generally occurs through ingestion of material contaminated with the toxin producing bacteria or absorbance of the toxin through the skin.
SNARE complexes containing SNAP are also targets for tetanus toxin which likewise inhibit vesicle fusion and neurotransmitter release through anterograde transport of the toxin into the CNS. Prevention of 20S SNARE complex assembly due to cleaving of substituent proteins prevents SNAPs from interacting with the receptor proteins in a non-competitive manner.
Genetics
Expression of the three SNAP proteins in mammalian is tissue dependent with α-SNAP (33kD) and γ-SNAP (36kD) expressed throughout the body, and β-SNAP (34kD) primarily found in brain tissues.[1] α-SNAP and β-SNAP share approximately 83% sequence homology and are encoded by NAPA and NAPB on chromosomes 19 and 18, respectively in humans.[7] β-SNAP protein is encoded by the NAPB on chromosome 20. Changes in temporal expression have been observed in rodent models during embryonic development but similar changes in humans is yet to be verified.[1] Expression data in the early years after discovery of the protein group in the 1990s were primarily confirmed though use of Western blott and allowed expression of the mRNA and later cDNA. Use of Immunofluorescent localization showed strong association of the proteins to intracellular membranes including the ER and Golgi bodies as well as vesicles.[8]
Deletions in α-SNAP gene have also been found to be lethal in utero in rodent models with hyh (hydrocephalus with hop gait)[9] while hyh due to missense mutations lead to 40% lower levels of expression.[10] The effects of the mutations develop in utero and become more severe over time, ultimately leading to worsening hydrocephalus and death. Reduced expression of α-SNAP in hyh/hyh mice is also associated with CD4 T-cell effector cytokine deficiency.[11]
Yeast (S. cerevisiae) homolog of the SNAP gene known as Sec17p has 67% similarity to mammalian α-SNAP[1] or approximately 34% homology with alpha and 33% with beta.[8] It has been studied based on its function in yeast vacuolar fusion.[2] The lethality of the double null mutation in this animal highlights the importance of this class of proteins in intra and inter-cell communication and survival.
Structure
Use of TEM and FRET imaging techniques was widely applied at the beginning of the century to resolve the SNARE complex and expanded to include SNAP proteins as well. The 20S complex ultimately forms a rod of 2.5 nm width by 15 nm in length that assembles along the axis of two coiled coils of interacting SNARE proteins.[4][12] The binding of SNAP to the lateral side of SNARE complex rod takes place at the membrane during the priming step. This interaction requires intact N-terminal residues 63 and 37 on the SNARE protein[12] which may directly interact with one or more alpha-helical domains of the SNAP. NSF binding to α-SNAP has also been shown to be negatively impacted by the phosphorylation of NSF or the Y83E mutant that displays phosphomimic properties.[13] The unwinding of the coiled-coil structures following ATP hydrolysis by NSF is also accompanied by a conformational change in syntaxin (SNARE) prior to vesicle fusion.
These structural finding have been confirmed by use of Quick-Freeze/Deep-Etch EM that also describes the ternary SNARE complex as a similarly elongated rod-like assembly around the SNARE proteins with N-terminal binding of SNAP.[14]
The yeast homolog Sec17, pictured above contains fourteen α-helices and has the approximate dimensions of 85 Å × 35 Å × 35 Å with multiple conserved residues along the packing face of the protein.[5] Blocking of Sec17/SNAP interaction with SNAREs and Sec18/NSF has recently been reported in the literature using small molecules binding to PA (phosphatidic acid) to prevent priming activity and limit vesicle fusion.[3]
Role in disease
Blocking of SNARE complex assembly, and therefore indirectly interfering with SNAP function, has a wide variety of application as evidenced by the diverse treatment utility of Botox which can be used to block vesicle fusion and neurotransmitter release. Targeting of SNAP protein receptors has been found both to be disease causing and has broad application when targeted with therapeutics.
Outlined below are recent publications indicating more direct associations of SNAPs in disease course and development. Notably, the role of SNAPs in disease states is still primarily related to its interaction as part of the SNARE complexes. Abnormal levels of multiple vesicular trafficking proteins are often observed in conjunction and a compound effect may lead to a disruption in signaling.
Colorectal cancer
In a studies of colorectal cancer of neuroendocrine markers, the expression of α-SNAP and β-SNAP were found to be higher in undifferentiated cells when compared to controls, and were associated with more aggressive disease.[15] Similarly, expression of other vesicle trafficking proteins including synaptophysin, SNAP-25 (SNARE), VAMP2 and syntaxin-1 were also found to have various levels of increase small cell undifferentiated carcinomas.[11]
Aberrant of signaling and trafficking of proteins in cancer cells has been previously reported based on SNARE complex interactions for α-SNAP within implication of its role as a negative regulator of autophagy and the MAPK pathway thorough dephosphorylating.[16] Depletion of α-SNAP has been reported to impair Golgi body integrity and assembly of vesicle fusion proteins at signaling junctions, while overexpression delays apoptosis in HeLa cells.[16]
Epilepsy
Association of α-SNAP with v-SNARE (vesicle), t-SNARE (target) proteins with synatxin-1 forms the 7S SNARE complex in central neurons used in vesicle transport.[10] Downregulation of alpha SNAPs has been documented to increase susceptibility to seizures in rodent models. In the same study a decrease alpha SNAP expression has been observed in patients with temporal lobe epilepsy as well as in the epileptic rat model.[17] An accumulation of the 7S complexes was also observed in synapse of the hippocampus in chronic rodent models for epilepsy.[18] The suspected mechanism may involve priming of the SNARE-SNAP-NSF complex to increase vesicle fusion at the membranes, however the exact mechanism by which the upregulation of the 7S complex occurs in not well understood.
Down syndrome
In a study of fetal brain development β-SNAP levels were found to be comparable between samples taken from Down syndrome (DS) affected and non-affected individuals. Presence of α-SNAP in comparison was only observed in half of DS affected samples.[19][7] Reduction in α-SNAP along with other observed changes to protein expression may indicate impaired synaptogenesis from very early on in development.
Huntington's disease
Vesicle fusion proteins evaluated in a study of rodent Huntington's disease (HD) model found higher levels of α-SNAP in the hippocampus and lower expression in the striatum of HD mice compared to controls.[20] It is notable that multiple other proteins involved in vesicle fusion also experienced decreased expression in the striatum along with increased expression in the hippocampus and the contributing effects could not yet be deconvoluted. The interaction of mutant huntingtin gene and vesicle fusion proteins may also be potentially responsible for deranged synaptic development or degeneration observed in the condition.[7]
Prion disease
Upregulation of α-SNAP was observed in mice with knock out 14-3-3 gamma protein suggesting a relationship between progression but not the pathogenesis of Creutzfeldt-Jakob Disease (CJD). Increased levels of 14-3-3 proteins are used diagnostically to confirm CJD but based on literature may not play a causal role in the disease.[21][7]
Intervention strategies
Interaction of α-SNAP with AMPA receptors for glutamate may be potential target to improve synaptic plasticity through mechanism of stabilization at membranes where SNAPs are present.[7] Additionally, α-SNAP has been implicated in surfactant and acrosomal exocytosis in alveolar cells and sperm cells respectively, although the exact mechanism are yet to be identified. SNAP protein isoforms are not a currently druggable target and may prove difficult to target as they serve primarily a scaffolding role. Insufficiency in expression is indicated in a number of neurodegenerative and immune related conditions where the primary treatment strategy may focus on gene-therapy as replacement option.
The potential for application to clinical therapy include the development of targeted regulators for β-SNAP for treatment of CNS pathologies including epilepsy.[1] Use of Inositol Polyphosphates to inhibit β-SNAP and synaptogamin interactions can also block neurotransmitter release and may be potentially useful in broader regulations of synaptic networks.
Small molecule agents that can be used to block SNARE complex activity through interaction with SNAPs and have been used in vitro,[3] but their practical use may extend to in vivo systems as well. In colorectal cancers where elevated α-SNAP levels were observed, siRNA technology may be employed to deplete overexpression,[7] but the novelty of this technology may be limited until further experience with the platform is gather and safety is well-demonstrated.
References
- 1 2 3 4 5 6 7 Stenbeck G (May 1998). "Soluble NSF-attachment proteins". The International Journal of Biochemistry & Cell Biology. 30 (5): 573–577. doi:10.1016/S1357-2725(97)00064-2. PMID 9693958.
- 1 2 Rizo J (August 2018). "Mechanism of neurotransmitter release coming into focus". Protein Science. 27 (8): 1364–1391. doi:10.1002/pro.3445. PMC 6153415. PMID 29893445.
- 1 2 3 Sparks RP, Arango AS, Starr ML, Aboff ZL, Hurst LR, Rivera-Kohr DA, et al. (November 2019). "A small-molecule competitive inhibitor of phosphatidic acid binding by the AAA+ protein NSF/Sec18 blocks the SNARE-priming stage of vacuole fusion". The Journal of Biological Chemistry. 294 (46): 17168–17185. doi:10.1074/jbc.RA119.008865. PMC 6873166. PMID 31515268.
- 1 2 3 Nichols BJ, Pelham HR (August 1998). "SNAREs and membrane fusion in the Golgi apparatus". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1404 (1–2): 9–31. doi:10.1016/S0167-4889(98)00044-5. PMID 9714710.
- 1 2 Rice LM, Brunger AT (July 1999). "Crystal structure of the vesicular transport protein Sec17: implications for SNAP function in SNARE complex disassembly". Molecular Cell. 4 (1): 85–95. doi:10.1016/S1097-2765(00)80190-2. PMID 10445030.
- ↑ Chen YA, Scheller RH (February 2001). "SNARE-mediated membrane fusion". Nature Reviews. Molecular Cell Biology. 2 (2): 98–106. doi:10.1038/35052017. PMID 11252968. S2CID 205012830.
- 1 2 3 4 5 6 Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA (October 2006). "A ubiquitous membrane fusion protein alpha SNAP: a potential therapeutic target for cancer, diabetes and neurological disorders?". Expert Opinion on Therapeutic Targets. 10 (5): 723–733. doi:10.1517/14728222.10.5.723. PMID 16981829. S2CID 26951791.
- 1 2 Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrow SA, Rothman JE (March 1993). "SNAP family of NSF attachment proteins includes a brain-specific isoform". Nature. 362 (6418): 353–355. Bibcode:1993Natur.362..353W. doi:10.1038/362353a0. PMID 8455721. S2CID 4341471.
- ↑ Chae TH, Kim S, Marz KE, Hanson PI, Walsh CA (March 2004). "The hyh mutation uncovers roles for alpha Snap in apical protein localization and control of neural cell fate". Nature Genetics. 36 (3): 264–270. doi:10.1038/ng1302. PMID 14758363. S2CID 798893.
- 1 2 Hong HK, Chakravarti A, Takahashi JS (February 2004). "The gene for soluble N-ethylmaleimide sensitive factor attachment protein alpha is mutated in hydrocephaly with hop gait (hyh) mice". Proceedings of the National Academy of Sciences of the United States of America. 101 (6): 1748–1753. doi:10.1073/pnas.0308268100. PMC 341847. PMID 14755058.
- 1 2 Miao Y, Bhushan J, Dani A, Vig M (May 2017). "Na+ influx via Orai1 inhibits intracellular ATP-induced mTORC2 signaling to disrupt CD4 T cell gene expression and differentiation". eLife. 6: e25155. doi:10.7554/eLife.25155. PMC 5459575. PMID 28492364.
- 1 2 Hohl TM, Parlati F, Wimmer C, Rothman JE, Söllner TH, Engelhardt H (November 1998). "Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes". Molecular Cell. 2 (5): 539–548. doi:10.1016/S1097-2765(00)80153-7. PMC 5496501. PMID 9844627.
- ↑ Morgan A, Burgoyne RD (November 2004). "Membrane traffic: controlling membrane fusion by modifying NSF". Current Biology. 14 (22): R968–R970. doi:10.1016/j.cub.2004.10.045. PMID 15556857. S2CID 11766249.
- ↑ Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE (August 1997). "Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy". Cell. 90 (3): 523–535. doi:10.1016/S0092-8674(00)80512-7. PMID 9267032. S2CID 13913393.
- ↑ Grabowski P, Schönfelder J, Ahnert-Hilger G, Foss HD, Heine B, Schindler I, et al. (September 2002). "Expression of neuroendocrine markers: a signature of human undifferentiated carcinoma of the colon and rectum". Virchows Archiv. 441 (3): 256–263. doi:10.1007/s00428-002-0650-9. PMID 12242522. S2CID 21987523.
- 1 2 Meng J, Wang J (August 2015). "Role of SNARE proteins in tumourigenesis and their potential as targets for novel anti-cancer therapeutics". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1856 (1): 1–12. doi:10.1016/j.bbcan.2015.04.002. PMID 25956199.
- ↑ Xi Z, Deng W, Wang L, Xiao F, Li J, Wang Z, et al. (November 2015). "Association of Alpha-Soluble NSF Attachment Protein with Epileptic Seizure". Journal of Molecular Neuroscience. 57 (3): 417–425. doi:10.1007/s12031-015-0596-4. PMID 26156199. S2CID 16026967.
- ↑ Matveeva EA, Vanaman TC, Whiteheart SW, Slevin JT (March 2007). "Asymmetric accumulation of hippocampal 7S SNARE complexes occurs regardless of kindling paradigm". Epilepsy Research. 73 (3): 266–274. doi:10.1016/j.eplepsyres.2006.11.003. PMC 1868484. PMID 17174072.
- ↑ Weitzdoerfer R, Dierssen M, Fountoulakis M, Lubec G (2001). "Fetal life in Down Syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure". In Lubec G (ed.). Protein Expression in Down Syndrome Brain. Vienna: Springer Vienna. pp. 59–70. doi:10.1007/978-3-7091-6262-0_5. ISBN 978-3-211-83704-7. PMID 11771761.
{{cite book}}:|journal=ignored (help) - ↑ Morton AJ, Faull RL, Edwardson JM (September 2001). "Abnormalities in the synaptic vesicle fusion machinery in Huntington's disease". Brain Research Bulletin. 56 (2): 111–117. doi:10.1016/S0361-9230(01)00611-6. PMID 11704347. S2CID 35996576.
- ↑ Steinacker P, Schwarz P, Reim K, Brechlin P, Jahn O, Kratzin H, et al. (February 2005). "Unchanged survival rates of 14-3-3gamma knockout mice after inoculation with pathological prion protein". Molecular and Cellular Biology. 25 (4): 1339–1346. doi:10.1128/MCB.25.4.1339-1346.2005. PMC 547999. PMID 15684385.