
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic.[1] This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.
Humans have an incredibly complex and intricate genome that has been shaped and modified over time by evolution. About 99.9% of the DNA-sequence in the human genome is conserved between individuals from all over the world, but some variation does exist.[1] Single nucleotide polymorphisms (SNPs) are considered to be the largest contributor to genetic variation in humans since they are so abundant and easily detectable.[2] It is estimated that there are at least 10 million SNPs within the human population but there are also many other types of genetic variants and they occur at dramatically different scales.[1] The variation between genomes in the human population range from single nucleotide polymorphisms to dramatic alterations in the human karyotype.[3]
Human genetic variation is responsible for the phenotypic differences between individuals in the human population. There are different types of genetic variation and it is studied extensively in order to better understand its significance. These studies lead to discoveries associating genetic variants to certain phenotypes as well as their implications in disease. At first, before DNA sequencing technologies, variation was studied and observed exclusively at a microscopic scale. At this scale, the only observations made were differences in chromosome number and chromosome structure. These variants that are about 3 Mb or larger in size are considered microscopic structural variants.[1] This scale is large enough to be visualized using a microscope and include aneuploidies, heteromorphisms, and chromosomal rearrangements.[1] When DNA sequencing was introduced, it opened the door to finding smaller and incredibly more sequence variations including SNPs and minisatellites. This also includes small inversions, duplications, insertions, and deletions that are under 1 kb in size.[1] In the human genome project the human genome was successfully sequenced, which provided a reference human genome for comparison of genetic variation. With improving sequencing technologies and the reference genome, more and more variations were found of several different sizes that were larger than 1 kb but smaller than microscopic variants. These variants ranging from about 1 Kb to 3 Mb in size are considered submicroscopic structural variants.[1] These recently discovered structural variants are thought to play a very significant role in phenotypic diversity and disease susceptibility.
Types of structural variants
Structural variation is an important type of human genetic variation that contributes to phenotypic diversity.[2] There are microscopic and submicroscopic structural variants which include deletions, duplications, and large copy number variants as well as insertions, inversions, and translocations.[1] These are several different types of structural variants in the human genome and they are quite distinctive from each other. A translocation is a chromosomal rearrangement, at the inter- or intra-chromosomal level, where a section of a chromosome changes position but with no change in the whole DNA content.[1] A section of DNA that is larger than 1 kb and occurs in two or more copies per haploid genome, in which the different copies share greater than 90% of the same sequence, are considered to be segmental duplications or low-copy repeats.[1] These are only a few of the several different types of structural variants that have been known to exist in the human genome. A table visualizing these different forms of structural variants, as well as others, is shown in Figure 1.
An inversion is a section of DNA on a chromosome that is reversed in its orientation in comparison to the reference genome.[1] There have been many studies identifying inversions because they have been found to have a big role in many diseases. A study found that forty percent of haemophilia A patients had a factor 8 gene inversion of a certain region that was four hundred kb in size.[4] The inversion breakpoint was found to be around a segmental duplication which is observed in many other inversion events.[4]
It is difficult to completely understand how each structural variant is created. It was previously known that repeated sequences on a chromosome increases the probability of non allelic homologous recombination.[5] These repeated sequences could cause deletions, duplications, inversions, and inverted duplication chromosomes. The products of this mechanism from the sequence repeats is depicted in Figure 2. A study was done on the olfactory receptor gene clusters where they questioned if there was an association between normal rearrangement of 8p and the repeated inverted sequences. The researchers observed that the rearrangement of chromosomes was actually caused by the homologous recombination in the 8p-reps. Therefore, they concluded that the substrate used in order to make rearrangements at the intrachromosomal level are the genes for olfactory receptors.[5] This discovery revealed the role that inverted duplicates have in affecting the development of structural variants. The mechanisms and ways in which structural variants are produced are important to better understand the development of these type of genetic variants.
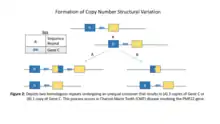
Copy-number variation
Copy-number variants are defined as sections of DNA that exist in a variable copy number when comparing it to the reference genome and are larger than 1 kb in size.[1] This definition is broad and includes deletions, duplications, and large copy number variants. If the copy number variant is present in 1% or more of the population then it is also considered a copy-number polymorphism.[1] There was a study on the global variation in copy number in the human genome which questioned the characteristics of copy number variants in the human genome. It was known that copy number variation in the human genome is important but at this point of time, it had not yet been fully understood. Human genome variation itself is very diverse as there are many types including inversions, duplication, SNPs, and other forms. They surveyed the genomes of 270 individuals, from a variety of populations, for copy number variants with technologies such as SNP arrays.[6] Their results showed that many copy number variants had specific arrangements of linkage disequilibrium which revealed the copy number variation in all of the different populations.[6] The study concluded that twelve percent of the genome contained CNVRs. They were found to be involved in more of the DNA in each genome than single nucleotide polymorphisms.[6] This was a remarkable discovery since single nucleotide polymorphisms have been known to be the greatest in number in the human genome. In terms of size, however, these type of structural variants were found to have a larger presence in the human genome.
The copy number variants continued to be studied as several studies continued to reveal the depth of their presence and their significance. A study was conducted that questioned the role of the organization of copy number variants and wondered what type of duplications they are. It was known that copy number variation plays a big role in many human diseases but at the time large scale studies of these duplications had not been done. They decided to sequence 130 breakpoints from 112 individuals that contained 119 known CNVs by doing whole genome sequencing as well as next generation sequencing.[7] They found that tandem duplications comprised 83% of the CNVs while 8.4% were triplications, 4.2% were adjacent duplications, 2.5% were insertional translocations, and 1.7% were other complex rearrangements.[7] The copy number variants were predominantly tandem duplications which made it the most common type of copy number variant in the human genome according to the results of the study on this population. More was needed on the mechanistic side of the formation of structural variants. There was a study that focused on the mechanisms of very interesting and rare pathogenic copy number variants. The researchers knew that copy number variation is important in genome structural variation and contributes to human genetic disease but the actual mechanisms of most of the new and few pathogenic copy number variants had not been known. They used sequencing technologies to sequence breakpoint areas of many rare pathogenic copy number variants which was the biggest and most in depth analysis of copy number variants. They saw that the genomic architectural features were very important in the human genome and they were associated with about eighty-one percent of breakpoints.[8] They concluded that tandem duplications and microdeletions that are rare and pathogenic do not happen in the human genome by chance. Instead, they arise from many different genomic architectural features.[8] It was a very interesting result in that the certain architectural features of the genome physically made it possible and probable to develop certain rare and pathogenic structural variants.
Structural variation can be seen as an avenue of genome modification for adaptation by evolution. A study was conducted on ancestral diet and the evolution of the human amylase gene copy number. The consumption of starch became a huge component of the human diet with the development of agricultural societies. Amylase is the enzyme that breaks down starch and its copy number varies.[9] These observations led to the question of whether or not the differences in starch consumption between different populations created natural selection pressures on the enzyme amylase. They tested for the differences in the amylase protein expression in saliva from different populations and compared their expression to their copy number in their respective genomes.[9] Then they compared the starch consumption of different populations to their copy number of the amylase gene. They found that there was more amylase protein expression in saliva from people that had higher amylase copy number in their genome and there was also an association between groups of people with high starch diets and a larger amylase gene copy number.[9] This study brought exciting results as structural variation proved an involvement in the evolution of the human population by increasing its amylase copy number over time.
The 1000 genomes project was able to successfully produce the DNA sequence of the human genome. They provided much sequencing data from many populations to analyze as well as a reference human genome for comparison and future studies. One study took advantage of this resource to question the structural variation differences between genomes from whole genome sequence data. It was known that human diseases are affected by duplications and deletions and that copy number analysis is common but multiallelic copy number variants (mCNVs) were not as well studied. The researchers got their data from the 1000 genomes project and analyzed 849 different genomes from a variety of populations that were sequenced in order to find large mCNVs.[10] From their analysis, they found that mCNVs create most genetic variation in gene dosage compared to other structural variants and that the gene expression variation is created by the dosage diversity of genes created by mCNVs.[10] The study underlined the great significance that structural variants, especially mCNVs, have on gene dosage which leads to variable gene expressions and human phenotypic diversity in the population.
Implications in disease
Charcot-Marie Tooth (CMT) disease
There are several structural variants in the human genome that have been observed but have not led to any obvious phenotypic effects.[1] There are some, however, that play a role in gene dosage which could lead to genetic diseases or distinct phenotypes. Structural variants can directly affect gene expression, such as with copy-number variants, or indirectly through position effects.[1] These effects can have significant implications in susceptibility to disease. The first gene dosage effect that was observed, and considered to be an autosomal dominant disease from an inherited DNA rearrangement, was Charcot-Marie Tooth (CMT) disease. Most of the associations found with CMT were with a 1.5 Mb tandem duplication in 17p11.2-p12 at the PMP22 gene.[11] The proposed mechanism for the structural variation is shown in Figure 2. When an individual has three copies of the normal gene, it results in the disease phenotype.[11] If the individual had only one copy of the PMP22 gene, on the other hand, the result was a clinically different hereditary neuropathy with liability to pressure palsies.[11] The differences in gene dosage created vastly different disease phenotypes which revealed the significant role that structural variation has on phenotype and susceptibility to disease.
HIV susceptibility
Structural variation studies became increasingly popular due to the discovery of their possible roles and effects in the human genome. Copy number variation is a very important type of structural variation and has been studied extensively. A study on the influence of the CCL3L1 gene on HIV-1/AIDS susceptibility tested if the copy number of the CCL3L1 gene had any effect on an individual’s susceptibility to HIV-1/AIDS. They sampled several different individuals and populations for their CCL3L1 copy number and compared it to their HIV acquirement risk. They found that there is an association between higher amounts in the copy number of CCL3L1 and susceptibility to HIV and AIDS since individuals who were more prone to HIV had a low copy number of CCL3L1.[12] This difference in copy number was shown to play a possibly significant role in HIV susceptibility due to this association. Another study that focused on the pathogenesis of human obesity tested if structural variation of the NPY4R gene was significant in obesity. Studies had previously shown that 10q11.22 CNV had an association with obesity and that several copy number variants were associated with obesity. Their CNV analysis revealed that the NPY4R gene had a much higher frequency of 10q11.22 CNV loss in the patient population.[13] The control population, on the other hand, had more CNV gain in the same region. This led the researchers to conclude that the NPY4R gene played an important role in the pathogenesis of obesity due to its copy number variation.[13] Studies involving copy number variation as well as other structural variants have brought new insights to the significant roles that structural variants play in the human genome.
Schizophrenia
The factors that contribute to the development of schizophrenia have been studied extensively. A very recent study was conducted on the mechanism and genes responsible for schizophrenia development. It had been previously shown that variation at an MHC locus was associated with the development of schizophrenia. This study found that the association is caused partly by the complement component 4 (C4) genes and therefore implying that allele variants of the C4 genes contribute to the development of schizophrenia.[14] Linkage disequilibrium helped researchers identify which C4 structural variant an individual had by looking at the SNP haplotypes. The SNP haplotypes and the C4 alleles were linked which was why they were in linkage disequilibrium, meaning that they segregated together.[14] A single structural C4 variant was associated with many different SNP haplotypes, but different SNP haplotypes where associated with only one C4 structural variant.[14] This was due to the linkage disequilibrium which allowed the researchers to determine the C4 structural variant easily by looking at the SNP haplotype. Their data suggested this because the results showed that the structural variants of C4 express the C4A protein at different levels and this difference in higher C4A protein expressions were associated with higher rates of schizophrenia development.[14] The different structural variant alleles of the same gene were shown to have different phenotypes and susceptibility to disease. These studies exhibit the breadth of the involvement and significance of structural variation on the human genome. Its importance is demonstrated with its contribution to phenotypic diversity and disease susceptibility.
Future directions
Many studies have been conducted to better understand human genome structural variation. There have been great advances in the research but its significance is still not fully understood. There are several questions still left unanswered which beg for further studies on the subject. Current studies usually target “unique” areas of the genome but are not able to detect the phenotypic effect of structural variants in highly repetitive, duplicated, and complex genomic areas.[15] It is very difficult to study this with the genomic technology of today but this may change with future development of sequencing technologies. In order to better understand the phenotypic effect of structural variants, large databases of genotypes and phenotypes of individuals must be created in order to make accurate associations. Huge projects such as Deciphering Developmental Disorders, UK10K, and International Standards for Cytogenomic Arrays Consortium have already paved the way to create databases for researchers to more easily pursue these studies.[15]
In addition, there has been growth and development in technology to create induced pluripotent stem cells with specific diseases. This introduces appropriate model systems to recreate disease causing structural variants such as translocations, duplications, and inversions.[15] The future advancement in technologies and large database efforts will help lead the way to better quality studies and a much better understanding of human genome structural variation.
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Feuk, L.; Carson, A.R.; Schere, S.W. (2006). "Structural variation in the human genome". Nature Reviews Genetics. 7 (2): 85–97. doi:10.1038/nrg1767. PMID 16418744. S2CID 17255998.
- 1 2 Nguyen, D.Q.; Webber, C.; Ponting, C.P. (2006). "Bias of selection on human copy-number variants". PLOS Genetics. 2 (2): e20. CiteSeerX 10.1.1.276.7715. doi:10.1371/journal.pgen.0020020. PMC 1366494. PMID 16482228.
- ↑ Kidd, J.M.; Cooper, G.M.; Donahue, W.F.; Hayden, H.S.; Sampas, N.; Graves, T.; Hansen, N.; Teauge, B.; Alkan, C.; Antonacci, F.; Haugen, E.; Zerr, T.; Yamada, N.A.; Tsang, P.; Newman, T.L.; Tuzun, E.; Cheng, Z.; Ebling, H.M.; Tusneem, N.; David, R.; Gillett, W.; Phelps, K.A.; Weaver, M.; Saranga, D.; Brand, A.; Tao, W.; Gustafson, E.; McKernan, K.; Chen, L.; Malig, M.; Smith, J.D.; Korn, J.M.; McCarroll, S.A.; Altshuler, D.A.; Peiffer, D.A.; Dorschner, M.; Stamatoyannopoulos, J.; Schwartz, D.; Nickerson, D.A.; Mullikin, J.C.; Wilson, R.K.; Bruhn, L.; Olson, M.V.; Kaul, R.; Smith, D.R.; Eichler, E.E. (2008). "Mapping and sequencing of structural variation from eight human genomes". Nature. 453 (7191): 56–64. Bibcode:2008Natur.453...56K. doi:10.1038/nature06862. PMC 2424287. PMID 18451855.
- 1 2 Lakich, D.; Kazazian, H.H.; Antonarakis, S.E.; Gitschier, J. (1993). "Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A". Nat. Genet. 5 (3): 236–241. doi:10.1038/ng1193-236. PMID 8275087. S2CID 25636383.
- 1 2 Giglio, S.; Broman, K.W.; Matsumoto, N.; Calvari, V.; Gimelli, G.; Neumann, T.; Ohashi, H.; Voullaire, L.; Larizza, D.; Giorda, R.; Weber, J.L.; Ledbetter, D.H.; Zuffardi, O. (2001). "Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements". American Journal of Human Genetics. 68 (4): 874–83. doi:10.1086/319506. PMC 1275641. PMID 11231899.
- 1 2 3 Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; Cho, E.K.; Dallaire, S.; Freeman, J.L.; Gonzalez, J.R.; Gratacos, M.; Huang, J.; Kalaitzopoulos, D.; Komura, D.; MacDonald, J.R.; Marshall, C.R.; Mei, R.; Montgomery, L.; Nishimura, K.; Okamura, K.; Shen, F.; Somerville, M.J.; Tchinda, J.; Valsesia, A.; Woodwark, C.; Yang, F.; Zhang, J.; Zerjal, T.; Zhang, J.; Armengol, L.; Conrad, D.F.; Estivill, X.; Tyler-Smith, C.; Carter, N.P.; Aburatani, H.; Lee, C.; Jones, K.W.; Schere, S.W.; Hurles, M.E. (2006). "Global variation in copy number in the human genome". Nature. 444 (7118): 444–454. Bibcode:2006Natur.444..444R. doi:10.1038/nature05329. PMC 2669898. PMID 17122850.
- 1 2 Newman, S.; Hermetz, K.E.; Weckselblatt, B.; Rudd, M.K. (2015). "Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints". American Journal of Human Genetics. 96 (2): 208–220. doi:10.1016/j.ajhg.2014.12.017. PMC 4320257. PMID 25640679.
- 1 2 Vissers, L.E.; Bhatt, S.S.; Janssen, I.M.; Xia, Z.; Lalani, S.R.; Pfundt, R.; Derwinska, K.; de Vries, B.B.; Gilissen, C.; Hoischen, A.; Nesteruk, M.; Wisniowiecka-Kowalnik, B.; Smyk, M.; Brunner, H.G.; Cheung, S.W.; van Kessel, A.G.; Veltman, J.A; Stankiewicz, P. (2009). "Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture". Human Molecular Genetics. 18 (19): 3579–3593. doi:10.1093/hmg/ddp306. PMID 19578123.
- 1 2 3 Perry, G.H.; Dominy, N.J.; Claw, K.G.; Lee, A. S.; Fiegler, H.; Redon, R.; Werner, J.; Villanea, F.A.; Mountain, J.L.; Misra, R.; Carter, N.P.; Lee, C.; Stone, A.C. (2007). "Diet and the evolution of human amylase gene copy number variation". Nature Genetics. 39 (10): 1256–60. doi:10.1038/ng2123. PMC 2377015. PMID 17828263.
- 1 2 Handsaker, R.E.; Van Doren, V.; Berman, J.R.; Genovese, G.; Kashin, S.; Boettger, L.M.; McCarroll, S.A. (2015). "Large multiallelic copy number variations in humans" (PDF). Nature Genetics. 47 (3): 296–303. doi:10.1038/ng.3200. PMC 4405206. PMID 25621458.
- 1 2 3 Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; Chakravarti, A.; Patel, P.I. (1991). "DNA duplication associated with Charcot-Marie-Tooth disease type 1A". Cell. 66 (2): 219–232. doi:10.1016/0092-8674(91)90613-4. PMID 1677316. S2CID 13155635.
- ↑ Gonzalez, E.; Kulkarni, H.; Bolivar, H.; Mangano, A.; Sanchez, R.; Catano, G.; Nibbs, R.J.; Freedman, B.I.; Quinones, M.P.; Bamshad, M.J.; Murthy, K.K.; Rovin, B.H; Bradley, W.; Clark, R.A.; Anderson, S.A.; O'Connell, J.; Agan, B.K.; Ahuja, S.S; Bologna, R.; Sen, L.; Dolan, M.J.; Ahuja, S.K. (2005). "The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility". Science. 307 (5714): 1434–40. Bibcode:2005Sci...307.1434G. doi:10.1126/science.1101160. PMID 15637236. S2CID 8815153.
- 1 2 Aerts, E.; Beckers, S.; Zegers, D.; Van Hoorenbeck, K.; Massa, G.; Verrijken, A.; Verhulst, S.L.; Van Gaal, L.F.; Van Hul, W. (2016). "CNV analysis and mutation screening indicate an important role for the NPY4R gene in human obesity". Obesity. 24 (4): 970–6. doi:10.1002/oby.21435. PMID 26921218.
- 1 2 3 4 Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; Genovese, G.; Rose, S.A.; Handsaker, R.E.; Daly, M.J.; Carroll, M.C.; Stevens, B.; McCarroll, S.A. (2016). "Schizophrenia risk from complex variation of complement component 4". Nature. 530 (7589): 177–83. Bibcode:2016Natur.530..177.. doi:10.1038/nature16549. PMC 4752392. PMID 26814963.
- 1 2 3 Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. (2013). "Phenotypic impact of genomic structural variation: insights from and for human disease". Nature Reviews Genetics. 14 (2): 125–138. doi:10.1038/nrg3373. PMID 23329113. S2CID 27469511.