| Variable surface glycoprotein | |
|---|---|
| Identifiers | |
| Organism | |
| Symbol | Tb927.5.4730 |
| Alt. symbols | Tb05.26C7.380 |
| Entrez | 3657576 |
| Other data | |
| Chromosome | 5: 1.41 - 1.41 Mb |
| Variant surface glycoprotein MITAT 1.2 | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Organism | |||||||
| Symbol | N/A | ||||||
| Alt. symbols | VSG 221 | ||||||
| UniProt | P26332 | ||||||
| |||||||
Variant surface glycoprotein (VSG) is a ~60kDa protein which densely packs the cell surface of protozoan parasites belonging to the genus Trypanosoma. This genus is notable for their cell surface proteins. They were first isolated from Trypanosoma brucei in 1975 by George Cross.[1] VSG allows the trypanosomatid parasites to evade the mammalian host's immune system by extensive antigenic variation. They form a 12–15 nm surface coat. VSG dimers make up ~90% of all cell surface protein and ~10% of total cell protein. For this reason, these proteins are highly immunogenic and an immune response raised against a specific VSG coat will rapidly kill trypanosomes expressing this variant. However, with each cell division there is a possibility that the progeny will switch expression to change the VSG that is being expressed. VSG has no prescribed biochemical activity.
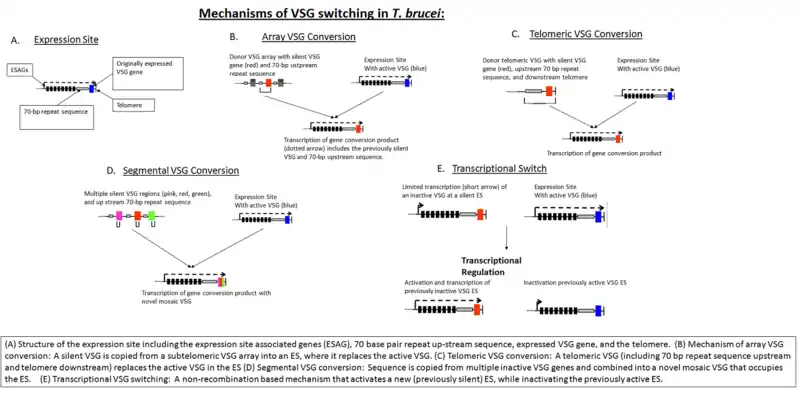
The parasite has a large cellular repertoire of antigenically distinct VSGs (~1500/2000 complete and partial (pseudogenes)) located in telomeric and subtelomeric arrays (on megabase chromosomes or minichromosomes). VSGs are expressed from a bloodstream expression site (BES, ES) in a polycistron by RNA polymerase I (recruited to a ribosomal-type promoter) with other ES-associated genes (ESAGs), of which transferrin receptor (Tfr: ESAG6, ESAG7) is one. Only one VSG gene is expressed at a time, as only one of the ~15 ES are active in a cell. VSG expression is 'switched' by homologous recombination of a silent basic copy gene from an array (directed by homology) into the active telomerically-located expression site.[2] During this transition, trypanosomes simultaneously display both pre- and post-switch VSGs on their surface. This coat replacement process is critical for the survival of recently switched cells because initial VSGs remain targets for the escalating host Ab response. Mosaic VSG genes can be created by homologous recombination of a partial VSG gene from an array. This partial gene may replace any portion of the residing VSG gene, creating a new mosaic VSG. VSG half-life measurements suggest that initial VSGs may persist on the surface of genetically switched trypanosomes for several days. It remains unclear whether the regulation of VSG switching is purely stochastic or whether environmental stimuli affect switching frequency. The fact that switching occurs in vitro suggests that there is at least some host-independent, stochastic element to the process.
The antigenic variation causes cyclical waves of parasitemia, which is one of the characteristics of human African trypanosomiasis. The cyclical process take 5–8 days. This occurs because a diverse range of coats expressed by the trypanosome population means that the immune system is always one step behind: it takes several days for an immune response against a given VSG to develop, giving the population time to diversify as individuals undergo further switching events. The repetition of this process prevents the extinction of the infecting trypanosome population, allowing chronic persistence of parasites in the host and enhancing opportunities for transmission.
In Trypanosoma brucei
In Trypanosoma brucei, the cell surface is covered by a dense coat of ~5 x 106 VSG dimers,[3] ~90% of all cell surface protein and ~10% of total cell protein.
The properties of the VSG coat that enable immune evasion are:
- Shielding – the dense nature of the VSG coat (VSG proteins pack shoulder-to-shoulder) prevents the immune system of the mammalian host from accessing the plasma membrane or any other parasitic invariant surface epitopes (such as ion channels, transporters, receptors etc.). The coat is uniform, made up of millions of copies of the same molecule; therefore, VSG is the only part of the trypanosome that the immune system can recognize.[4]
- Periodic antigenic variation – the VSG coat undergoes frequent stochastic genetic modification—'switching'—allowing variants expressing a new VSG coat to escape the specific immune response raised against the previous coat. This antigenic variation creates cyclical waves of parasitemia characteristic of Human African Trypanosomiasis.[5]
- Antigen 'cleaning' and VSG recycling—VSG is efficiently recycled through the trypanosome flagellar pocket, allowing antibodies to be 'cleaned' from VSG before re-incorporation back into the cellular membrane. Importantly, VSGs recognized and bound by antibodies are selectively pushed toward the flagellar pocket at a quicker rate than unidentified VSG; in this scenario, the antibody acts as a 'sail', which quickens the process of VSG being brought to the area of recycling.[6]
The VSGs from T. brucei are attached to the plasma membrane via a covalent attachment to two glycosyl-phosphatidylinositol (GPI) anchors (one per monomer),[7] which directs its forward-trafficking from the ER to the flagellar pocket for incorporation into the membrane, as predicted by the GPI valence hypothesis.[8][9]
VSGs are replaced by an equally dense coat of procyclins when the parasite differentiates into the procyclic form in the tsetse fly midgut. There is a very fast inhibition of VSG gene transcription which occurs as soon as the temperature is lowered.[10]
Expression
The source of VSG variability during infection is a large 'archive' of VSG genes present in the T. brucei genome. Some of these are full-length, intact genes; others are pseudogenes (typically with frameshift mutations, premature stop codons, or fragmentation).[11] Expression of an antigenically different VSG can occur by simply switching to a different full-length VSG gene by Expression Site switching (switching which ES is active). In addition, chimeric or 'mosaic' VSG genes can be generated by combining segments from more than one silent VSG gene. The formation of mosaic VSGs allows the (partial) expression of pseudogene VSGs, which can constitute the major portion of the VSG archive, and can contribute directly to antigenic variation, vastly increasing the trypanosome's capacity for immune evasion and posing a major problem for vaccine development.[12]
VSG genes can be kept silent and switched on at any given time. The expressed VSG is always located in an Expression Site (ES), which are specialised expression loci found at the telomeres of some of the large and intermediate chromosomes. Each ES is a polycistronic unit, containing a number of Expression Site-Associated Genes (ESAGs) all expressed along with the active VSG. While multiple ES exist, only a single one is ever active at one time. A number of mechanisms appear to be involved in this process, but the exact nature of the silencing is still unclear.[13]
The expressed VSG can be switched either by activating a different expression site (and thus changing to express the VSG in that site), or by changing the VSG gene in the active site to a different variant. The genome contains many copies of VSG genes, both on minichromosomes and in repeated sections in the interior of the chromosomes. These are generally silent, typically with omitted sections or premature stop codons, but are important in the evolution of new VSG genes. It is estimated up to 10% of the T.brucei genome may be made up of VSG genes or pseudogenes. Any of these genes can be moved into the active site by recombination for expression. Again, the exact mechanisms that control this are unclear, but the process seems to rely on DNA repair machinery and a process of homologous recombination.[14]
The bloodstream expression site (BES), or telomeric expression site, is used for exchanging variant surface glycoproteins when in host's blood stream to escape the complement system. BESs are polymorphic in size and structure but reveal a surprisingly conserved architecture in the context of extensive recombination. Very small BESs do exist and many functioning BESs do not contain the full complement of expression site associated genes (ESAGs).[15] There is a collection of an estimated 20-30 sites, each being active at a time.[16] Active VSG expression sites are depleted of nucleosomes.[17]
The gene repertoires in T. brucei have diverged to become strain-specific.[18]
The variant surface glycoprotein genes of T. brucei have been classified into two groups depending upon whether or not duplication of the genes is observed when they are expressed.[19]
Secretory trafficking
Trypanosoma have a simple, polarized membrane transport system consisting of a single ER, lysosome, and Golgi apparatus.
VSG is first transcribed as a polycistron and then undergoes trypanosomatid-specific poly-adenylation and trans-splicing directed by polypyrimidine tracts. Because there is no transcriptional control, the VSG 3'UTR is important for its RNA stability (most importantly, the 8mer and 14mer). VSG is then transcribed on membrane-bound polysomes, and the appearance of the N-terminal signal sequence directs VSG to the ER. VSG is thereby co-translationally transported into the ER lumen, rapidly N-glycosylated (on asn-x-ser/thr sites) and GPI anchored at the ω site by a transamination reaction (removing of the C-term hydrophobic 17 or 23 aa GPI anchoring sequence). The ω site is always Ser (usually in 17 aa signal sequence peptides), Asp (usually in 23 aa signal sequence peptides), or Asn. Also, the number of N-glycosylation sites per VSG may vary (usually 1-3 N-glycans). VSG MITat.1.5 is glycosylated at all three potential N-glycosylation sites.[20]
VSG then undergoes the calreticulin/calnexin folding cycle (calnexin is absent in Trypanosoma brucei), where it is transiently monoglucosylated and deglucosylated, and interacts with various ER chaperone proteins, such as BiP, in order to fold correctly. VSG efficiently folds and dimerizes (suggesting intrinsically favorable folding) and is transported through the Golgi to the flagellar pocket for incorporation into the cell membrane.
Importantly, following incorporation into the cellular membrane, VSG may later be recycled through the flagellar pocket and sorted back to the cell surface. VSG is not turned over by lysosomal or proteasomal canonical degradation pathways,[21] but is instead lost from the cell by specific cleavage of its GPI anchor by GPI-specific PLC.
Structure
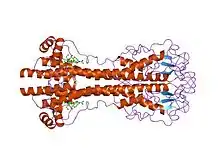
VSG genes are hugely variable at the sequence (primary) level, but variants are thought to have strongly conserved structural (tertiary) features, based on two determined 3-dimensional structures[22] and conservation of 2-dimensional sequence motifs (descending and ascending alpha-helices that make up the dimerization interface), allowing them to perform a similar shielding function.[23] VSGs are made up of N terminal domain of around 300–350 amino acids with low sequence homology (13–30% identity), and a more conserved C terminal domain of ~100 amino acids. N-terminal domains are grouped into classes A-C depending on their cysteine patterns. C-term domains are grouped by sequence homology into classes I-III, with apparently no restriction on which N-term classes they can pair with to form a full VSG. To dimerize, VSG N-terminal domains form a bundle of four alpha helices directed by hydrophobic interactions, around which hang smaller structural features (five smaller helices and three beta-sheets).
VSG is anchored to the cell membrane via a glycophosphatidylinositol (GPI) anchor—a noncovalent linkage from the C-terminus which directs its forward trafficking from the ER to the membrane. This GPI anchor is specifically cleaved by GPI Phospholipase C, cleaving the membrane-form VSG, and allowing VSG protein and portion of the GPI anchor to be lost into the extracellular milieu as soluble VSG (sVSG, which is can be recognized as Cross-Reacting Determinant, or CRD), while retaining the two 1,2-dimyristolglycerol chains in the membrane.
Antigenic variation
VSG is highly immunogenic, and an immune response raised against a specific VSG coat will rapidly kill trypanosomes expressing this variant. Antibody-mediated trypanosome killing can also be observed in vitro by a complement-mediated lysis assay. However, with each cell division there is a possibility that one or both of the progeny will switch expression to change the VSG that is being expressed. The frequency of VSG switching has been measured to be approximately 0.1% per division,[24] though switching rates do differ in culture vs. in vivo. As T. brucei populations can peak at a size of 1011 within a host[25] this rapid rate of switching ensures that the parasite population is constantly diverse. A diverse range of coats expressed by the trypanosome population means that the immune system is always one step behind: it takes several days for an immune response against a given VSG to develop, giving the population time to diversify as individuals undergo further switching events. Reiteration of this process prevents extinction of the infecting trypanosome population, allowing chronic persistence of parasites in the host, enhancing opportunities for transmission. The clinical effect of this cycle is successive 'waves' of parasitaemia (trypanosomes in the blood).[3]
In other trypanosomes
Variable surface glycoproteins are also found in other Trypanosoma species.
In Trypanosoma equiperdum, a parasite causing the covering sickness in horses, These proteins allow the parasite to efficiently evade the host animal's immune system.[26] These VSGs allow the organism to constantly manipulate and change the surface structure of its proteins, which means it is constantly being presented to the immune system as a new foreign organism and this prevents the body from mounting a large enough immune response to eradicate the disease.[26] In this sense, Trypanosoma equiperdum is a very efficient organism; it may infect fewer species than other diseases, but it infects and survives very efficiently within its specified hosts. The VSG proteins in T. equiperdum are also phosphorylated.[27]
A VSG gene from Trypanosoma evansi, a parasite that causes a form of surra in animals, has been cloned in Escherichia coli. The expressed protein is immunoreactive with all the sera combinations. The animals immunized with whole cell lysate or recombinant protein show similar antibody reactions in ELISA (enzyme-linked immunosorbent assay) and CATT (card agglutination test for trypanosomiasis).[28] The variable surface glycoprotein RoTat 1.2 PCR can be used as a specific diagnostic tool for the detection of T. evansi infections.[29]
The smallest VSG protein (40 kDa in size) to date (1996) has been found in Trypanosoma vivax, which bears little carbohydrate.[30]
In Trypanosoma congolense, in vitro analyses of the incorporated sugars after hydrolysis of the glycoprotein showed that glucosamine and mannose are utilized in the biosynthesis of the carbohydrate moiety directly whereas galactose was converted possibly to other intermediates before being incorporated into the antigen. The unglycosylated VSG with a molecular weight of 47 kDa had completely lost its size heterogeneity.[31]
See also
- Amastin, another surface (trans-membrane) glycoprotein in trypanosomatid parasites[32]
- Coat protein (disambiguation)
- Glycocalyx
- List of MeSH codes (D23)
- List of MeSH codes (D12.776.395)
- List of MeSH codes (D12.776.543)
References
- ↑ Cross GA (1975). "Identification, purification and properties of clone-specific glycoprotein antigens constituting the surface coat of Trypanosoma brucei". Parasitology. 71 (3): 393–417. doi:10.1017/s003118200004717x. PMID 645. S2CID 20749130.
- ↑ Buck GA, Jacquemot C, Baltz T, Eisen H (December 1984). "Re-expression of an inactivated variable surface glycoprotein gene in Trypanosoma equiperdum". Gene. 32 (3): 329–36. doi:10.1016/0378-1119(84)90008-8. PMID 6530143.
- 1 2 Barry JD, McCulloch R (2001). "Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite". Advances in Parasitology Volume 49. Vol. 49. pp. 1–70. doi:10.1016/S0065-308X(01)49037-3. ISBN 978-0-12-031749-3. PMID 11461029.
{{cite book}}:|journal=ignored (help) - ↑ Overath P, Chaudhri M, Steverding D, Ziegelbauer K (February 1994). "Invariant surface proteins in bloodstream forms of Trypanosoma brucei". Parasitology Today. 10 (2): 53–8. doi:10.1016/0169-4758(94)90393-X. PMID 15275499.
- ↑ Ross R, Thomson D (June 1910). "A Case of Sleeping Sickness showing Regular Periodical Increase of the Parasites Disclosed". British Medical Journal. 1 (2582): 1544–5. doi:10.1136/bmj.1.2582.1544. PMC 2331906. PMID 20765166.
- ↑ Rudenko G (2011-10-24). "African trypanosomes: the genome and adaptations for immune evasion". Essays in Biochemistry. 51: 47–62. doi:10.1042/bse0510047. PMID 22023441.
- ↑ Grab DJ, Verjee Y. "Localization of a Variable Surface Glycoprotein Phosphatidylinositol-Specific Phospholipase-C in Trypanosoma brucei brucei". FAO Corporate document depository. Food and Agricultural Organization of the United Nations.
- ↑ Triggs VP, Bangs JD (February 2003). "Glycosylphosphatidylinositol-dependent protein trafficking in bloodstream stage Trypanosoma brucei". Eukaryotic Cell. 2 (1): 76–83. doi:10.1128/ec.2.1.76-83.2003. PMC 141176. PMID 12582124.
- ↑ Schwartz KJ, Peck RF, Tazeh NN, Bangs JD (December 2005). "GPI valence and the fate of secretory membrane proteins in African trypanosomes". Journal of Cell Science. 118 (Pt 23): 5499–511. doi:10.1242/jcs.02667. PMID 16291721.
- ↑ Pays E, Coquelet H, Pays A, Tebabi P, Steinert M (September 1989). "Trypanosoma brucei: posttranscriptional control of the variable surface glycoprotein gene expression site". Molecular and Cellular Biology. 9 (9): 4018–21. doi:10.1128/mcb.9.9.4018. PMC 362464. PMID 2779574.
- ↑ Marcello L, Barry JD (September 2007). "Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure". Genome Research. 17 (9): 1344–52. doi:10.1101/gr.6421207. PMC 1950903. PMID 17652423.
- ↑ Barbour AG, Restrepo BI (2000). "Antigenic variation in vector-borne pathogens". Emerging Infectious Diseases. 6 (5): 449–57. doi:10.3201/eid0605.000502. PMC 2627965. PMID 10998374.
- ↑ Pays E (November 2005). "Regulation of antigen gene expression in Trypanosoma brucei". Trends in Parasitology. 21 (11): 517–20. doi:10.1016/j.pt.2005.08.016. PMID 16126458.
- ↑ Morrison LJ, Marcello L, McCulloch R (December 2009). "Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity" (PDF). Cellular Microbiology. 11 (12): 1724–34. doi:10.1111/j.1462-5822.2009.01383.x. PMID 19751359. S2CID 26552797.
- ↑ Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, Heath P, Kartvelishvili M, Mungall K, Harris D, Hauser H, Sanders M, Saunders D, Seeger K, Sharp S, Taylor JE, Walker D, White B, Young R, Cross GA, Rudenko G, Barry JD, Louis EJ, Berriman M (2008). "Telomeric expression sites are highly conserved in Trypanosoma brucei". PLOS ONE. 3 (10): e3527. Bibcode:2008PLoSO...3.3527H. doi:10.1371/journal.pone.0003527. PMC 2567434. PMID 18953401.
- ↑ Vanhamme L, Lecordier L, Pays E (May 2001). "Control and function of the bloodstream variant surface glycoprotein expression sites in Trypanosoma brucei". International Journal for Parasitology. 31 (5–6): 523–31. doi:10.1016/S0020-7519(01)00143-6. PMID 11334937.
- ↑ Stanne TM, Rudenko G (January 2010). "Active VSG expression sites in Trypanosoma brucei are depleted of nucleosomes". Eukaryotic Cell. 9 (1): 136–47. doi:10.1128/EC.00281-09. PMC 2805301. PMID 19915073.
- ↑ Hutchinson OC, Picozzi K, Jones NG, Mott H, Sharma R, Welburn SC, Carrington M (July 2007). "Variant Surface Glycoprotein gene repertoires in Trypanosoma brucei have diverged to become strain-specific". BMC Genomics. 8: 234. doi:10.1186/1471-2164-8-234. PMC 1934917. PMID 17629915.
- ↑ Young JR, Turner MJ, Williams RO (1984). "The role of duplication in the expression of a variable surface glycoprotein gene of Trypanosoma brucei". Journal of Cellular Biochemistry. 24 (3): 287–95. doi:10.1002/jcb.240240309. PMID 6736139. S2CID 73535.
- ↑ Mehlert A, Bond CS, Ferguson MA (October 2002). "The glycoforms of a Trypanosoma brucei variant surface glycoprotein and molecular modeling of a glycosylated surface coat". Glycobiology. 12 (10): 607–12. doi:10.1093/glycob/cwf079. PMID 12244073.
- ↑ Tiengwe C, Muratore KA, Bangs JD (November 2016). "Surface proteins, ERAD and antigenic variation in Trypanosoma brucei". Cellular Microbiology. 18 (11): 1673–1688. doi:10.1111/cmi.12605. PMC 5575760. PMID 27110662.
- ↑ Freymann D, Down J, Carrington M, Roditi I, Turner M, Wiley D (1990). "2.9 Å resolution structure of the N-terminal domain of a variant surface glycoprotein from Trypanosoma brucei". Journal of Molecular Biology. 216 (1): 141–60. doi:10.1016/S0022-2836(05)80066-X. PMID 2231728.
- ↑ Blum ML, Down JA, Gurnett AM, Carrington M, Turner MJ, Wiley DC (April 1993). "A structural motif in the variant surface glycoproteins of Trypanosoma brucei". Nature. 362 (6421): 603–9. Bibcode:1993Natur.362..603B. doi:10.1038/362603a0. PMID 8464512. S2CID 4370099.
- ↑ Turner CM (August 1997). "The rate of antigenic variation in fly-transmitted and syringe-passaged infections of Trypanosoma brucei". FEMS Microbiology Letters. 153 (1): 227–31. doi:10.1111/j.1574-6968.1997.tb10486.x. PMID 9252591.
- ↑ Barry JD, Hall JP, Plenderleith L (September 2012). "Genome hyperevolution and the success of a parasite". Annals of the New York Academy of Sciences. 1267 (1): 11–7. Bibcode:2012NYASA1267...11B. doi:10.1111/j.1749-6632.2012.06654.x. PMC 3467770. PMID 22954210.
- 1 2 Raibaud A, Gaillard C, Longacre S, Hibner U, Buck G, Bernardi G, Eisen H (July 1983). "Genomic environment of variant surface antigen genes of Trypanosoma equiperdum". Proceedings of the National Academy of Sciences of the United States of America. 80 (14): 4306–10. Bibcode:1983PNAS...80.4306R. doi:10.1073/pnas.80.14.4306. PMC 384026. PMID 6308614.
- ↑ Baltz T, Giroud C, Baltz D, Duvillier G, Degand P, Demaille J, Pautrizel R (1982). "The variable surface glycoproteins of Trypanosoma equiperdum are phosphorylated". The EMBO Journal. 1 (11): 1393–8. doi:10.1002/j.1460-2075.1982.tb01328.x. PMC 553222. PMID 6821334.
- ↑ Sengupta PP, Balumahendiran M, Balamurugan V, Rudramurthy GR, Prabhudas K (June 2012). "Expressed truncated N-terminal variable surface glycoprotein (VSG) of Trypanosoma evansi in E. coli exhibits immuno-reactivity". Veterinary Parasitology. 187 (1–2): 1–8. doi:10.1016/j.vetpar.2012.01.012. PMID 22277627.
- ↑ Claes F, Radwanska M, Urakawa T, Majiwa PA, Goddeeris B, Büscher P (September 2004). "Variable Surface Glycoprotein RoTat 1.2 PCR as a specific diagnostic tool for the detection of Trypanosoma evansi infections". Kinetoplastid Biology and Disease. 3 (1): 3. doi:10.1186/1475-9292-3-3. PMC 521498. PMID 15377385.
- ↑ Gardiner PR, Nene V, Barry MM, Thatthi R, Burleigh B, Clarke MW (November 1996). "Characterization of a small variable surface glycoprotein from Trypanosoma vivax". Molecular and Biochemical Parasitology. 82 (1): 1–11. doi:10.1016/0166-6851(96)02687-4. PMID 8943146.
- ↑ Reinwald E, Heidrich C, Risse HJ (May 1984). "In vitro studies on the biosynthesis of the surface glycoprotein of Trypanosoma congolense". The Journal of Protozoology. 31 (2): 300–6. doi:10.1111/j.1550-7408.1984.tb02966.x. PMID 6470988.
- ↑ Jackson AP (January 2010). "The evolution of amastin surface glycoproteins in trypanosomatid parasites". Molecular Biology and Evolution. 27 (1): 33–45. doi:10.1093/molbev/msp214. PMC 2794310. PMID 19748930.
External links
- Variant Surface Glycoproteins, Trypanosoma at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- www.icp.ucl.ac.be