Linked-read sequencing, a type of DNA sequencing technology, uses specialized technique that tags DNA molecules with unique barcodes before fragmenting them. Unlike traditional sequencing technology, where DNA is broken into small fragments and then sequenced individually, resulting in short read lengths that has difficulties in accurately reconstructing the original DNA sequence, the unique barcodes of linked-read sequencing allows scientists to link together DNA fragments that come from the same DNA molecule. A pivotal benefit of this technology lies in the small quantities of DNA required for large genome information output, effectively combining the advantages of long-read and short-read technologies.[1]
History
This sequencing method was originally developed by 10x Genomics in 2015, and was launched under the name 'GemCode' or 'Chromium'. GemCode employed a method of gel bead-based barcoding to amalgamate short DNA fragments.[2] The longer fragments produced by this could then be sequenced using validated technology such as Illumina next-generation sequencing.[2][3] An updated version of linked-read sequencing was introduced by the same company in 2018, termed 'Linked-Reads V2'. While GemCode uses a single barcode for tagging of both the gel bead and the DNA fragment, Linked-Reads V2 uses separate barcodes for improved detection of genetic variants.
The group developed the linked-read sequencing technology published their first paper regarding this technology in 2016. The authors of this paper developed the linked-read sequencing technology initially to sequence the genomes of both healthy individuals and cancer patients to determine somatic mutations, copy number variations, and structural variations in cancer genomes.[2] Later that year, another research group combined linked-read sequencing technology with long-read sequencing technology to assemble human genome.[3] Both studies demonstrated the utility of linked-read sequencing in comprehensive genome analysis and in understanding genetic diseases. However, in 2019, a lawsuit relating to patent infringement resulted in 10x Genomics discontinuing their line of linked-read products.
Method
Overview
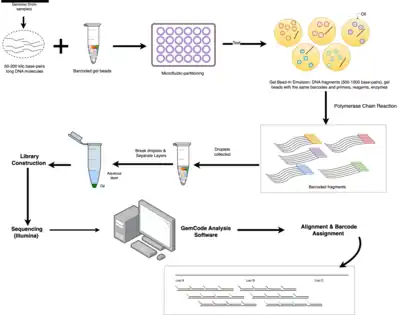
The linked-read sequencing is microfluidic-based, and only needs nanograms of input DNA.[2] One nanogram of DNA can be distributed across more than 100,000 droplet partitions, where DNA fragments are barcoded and subjected to polymerase chain reactions (PCR).[2] As a result, DNA fragments (or reads) that share the same barcode can be grouped as coming from one single long input DNA sequence.[2] And, long range information can be assembled from short reads.
Steps of Linked-read sequencing:[2]
- Sample Preparation: DNA is extracted from a sample (e.g., blood) and cut into fragments of 50 to 200 kilo base-pairs long.
- Barcode Sequencing: each DNA fragment is labelled with a unique barcode through a process known as "Gel Bead-In Emulsion" (GEM).
- Library Preparation: barcoded DNA fragments are amplified with PCR to generate sequencing libraries.
- Sequencing: with Illumina next-generation sequencing technology, generate millions to billions of short sequence reads that represent fragments of the original DNA molecules.
- Barcode Processing: group short reads to longer fragments based on barcodes.
- Downstream Analysis: processed reads are aligned to a reference genome, or used for de novo assembly of complex genomes, haplotype phasing, or identification of structural variations.
Barcode Sequencing
During barcode sequencing, high molecular weight DNA samples that contain the targeted DNA sequence, ranging from fifty to several hundred kilobases in size, are combined with gel beads containing unique barcodes, enzymes, and sequencing reagents.[2] Microfluidic device can partition input DNA molecules into individual nanoliter-sized droplets of water-in-oil emulsion, called GEMs.[2] Each GEM contains gel beads coated with the same barcode and primers, and a small amount of DNA.[2] The primers are complementary to specific regions of the DNA molecule, allowing for amplification of the DNA in the droplets through PCR.[2] The barcodes enable the identification and grouping of sequencing reads that originate from the same long fragment, which is crucial for downstream analysis.[2]
Library Preparation and Sequencing
The barcoded DNA fragments are amplified using PCR to create a library of DNA fragments with identical barcodes. All the fragments derived from a given DNA molecule are tagged with the same barcode.[4] This step increases the quantity of DNA for sequencing and reduces the chances of losing unique DNA fragments during sequencing. Droplets (or GEM) are later collected in a tube, and the emulsion is broken, releasing the amplified, barcoded DNA sequences.
Standard Illumina next-generation sequencing technology can be used to sequence libraries.[5] During sequencing, the barcodes are read along with the DNA sequences, allowing researchers and scientists to group together DNA fragments that originate from the same DNA molecule.[5] Even though each DNA fragment is typically not fully sequenced, the information from many overlapping fragments in the same genomic region can be combined to reconstruct the long stretches of the genome.[5] Therefore, a genome can be easily assembled from scratch without any prior reference.
Processing
The raw sequencing data is then processed through bioinformatics (e.g., the GemCode analysis software developed by 10x Genomics) to remove low-quality reads and to assign reads to their respective barcodes.[2] Reads can be aligned to a reference genome or assembled de novo to generate long-range contigs. The read alignment step is important for determining the order and orientation of the long DNA fragments, and for identifying genomic variations, such as insertions or deletions.
Applications
De Novo Genome Assembly
Linked-read sequencing can facilitate de novo genome assembly, which involves reconstructing a genome from scratch without any prior reference. Linked-read sequencing enables assembly of large genomic regions, and helps improve the completeness and contiguity of the resulting genome. This can be particularly useful for studying organisms that lack a high-quality reference genome, such as non-model organisms or organisms with complex genomes.[6] Many scientists have been using linked-read sequencing technology for de novo genome assembly recently in a variety of organisms, including humans, plants, and animals.[7][6][8] For example, Dr. Evan Eichler and his research group used linked-read sequencing to assemble genome of orangutan, which had previously been difficult to study due to its complex genome.[8] The resulting genome assembly helped scientists to study new insights into the evolutionary history of primates and the genetic basis of human diseases.[8] Also, the aligned or assembled reads can be used for other genetic investigations or downstream analysis, such as haplotype phasing.
Haplotype Phasing
Haplotype refers to a group of genetic variants inherited together on a chromosome from one parent due to their genetic linkage. Haplotype phasing (also called haplotype estimation) refers to the process of reconstructing individual haplotypes, important for determining the genetic basis of diseases.[9] Linked-read sequencing allows consistent coverage of genes related to different diseases, helping scientists to obtain all the regions carrying mutations from targeted genes.[10] For example, in 2018, a group of researchers used linked-read sequencing technology to sequence genetic information from a pregnant woman who was a carrier of Duchenne muscular dystrophy (DMD) mutation.[10] Linked-read sequencing allows them to identify the maternal haplotypes and determine the presence of the mutant alleles in the foetal DNA.[10] This non-invasive prenatal diagnosis of DMD demonstrates the clinical applicability of linked-read sequencing.
Structural Variation Analysis
Structural variations, such as deletions, duplications, inversions, translocations, and other rearrangements, are common in human genomes.[4] These variations can have significant impacts on genome functions, and have been implicated in many diseases. Linked-read sequencing technology labels all reads that originate from the same long DNA fragment with the same barcode, so it enables the detection of a large number of structural variants.[4] Complexity of structural variants can be resolved with linked-read sequencing, and provide a complete picture of the genomic landscape. Many scientists have already been using linked-read sequencing to identify and characterise structural variants in diverse populations, including people with genetic disorders or cancers [11]
Transcriptome Analysis
Transcriptome analysis is the study of all the RNA transcripts that are produced by the genome of an organism. Linked-read sequencing has been used by researchers to assemble transcript isoforms and alternative splicing events.[12] Information regarding alternative splicing events can provide insights into the regulation of gene expression in human transcriptome [12]
Epigenetic Analysis
Epigenetics refers to the study of heritable changes in genetic activities that are distinct from changes in DNA sequences. Epigenetic analysis involves studying DNA-protein interactions, histone modifications, and DNA methylation. Linked-read sequencing has been used for studying DNA methylation patterns by many studies.[13][14] For example, in 2021, a study investigated the DNA methylation differences in peripheral blood cells between twins, in which one twin had Alzheimer’s Disease and the other was cognitively normal.[13] Linked-read sequencing technology allowed researchers to identify more than 3000 differentially methylated regions between these twins discordant for Alzheimer’s Disease, and investigation of these differentially methylated regions eventually led to identification of genes enriched in neurodevelopmental processes, neuronal signalling, and immune system functions [13]
Use
Advantages
- Wide range of genomic applications and scientific questions, including de novo genome assembly, haplotype phasing, structural variant analysis, and transcriptome and epigenetic analysis.
- Accuracy and scalability.
- Method requires small quantities of input DNA, which can be beneficial for small samples or single cell studies.[2]
- More cost effective per sample in comparison with long-read technologies such as Oxford Nanopore sequencing.[3]
- Libraries produced by linked-read can be processed using Illumina short read sequencing, increasing accessibility.[2][3]
Limitations
- Complexity of library construction - this technology requires high molecular DNA preparation in order to produce long enough DNA molecules for sequencing.[3]
- Limitations in read length may result in limited haplotype resolution, which could reduce the efficacy of this technology in highly complex genomic regions.[2][3]
Controversy
In 2018, Bio-Rad Laboratories filed a lawsuit against 10x Genomics stating that their linked-read technology infringed on three patents which had been licensed from Bio-Rad at the University of Chicago.[15] Bio-Rad was awarded a sum of $23,930,716 by a jury. The 10x Genomics filed a motion for judgement as a matter of law (JMOL) but were denied in 2019, and the court proceedings concluded in 2020. Following this lawsuit, 10x Genomics discontinued their linked-read assay.[15] An exception was made for linked-read products which had already been sold by the company prior to the lawsuit, allowing 10x Genomics to continue to provide those researchers with services such as support and warranty maintenance for this technology.
References
- ↑ Marks, Patrick; Garcia, Sarah; Barrio, Alvaro Martinez; Belhocine, Kamila; Bernate, Jorge; Bharadwaj, Rajiv; Bjornson, Keith; Catalanotti, Claudia; Delaney, Josh; Fehr, Adrian; Fiddes, Ian T.; Galvin, Brendan; Heaton, Haynes; Herschleb, Jill; Hindson, Christopher (April 2019). "Resolving the full spectrum of human genome variation using Linked-Reads". Genome Research. 29 (4): 635–645. doi:10.1101/gr.234443.118. ISSN 1088-9051. PMC 6442396. PMID 30894395.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Zheng, Grace X. Y.; Lau, Billy T.; Schnall-Levin, Michael; Jarosz, Mirna; Bell, John M.; Hindson, Christopher M.; Kyriazopoulou-Panagiotopoulou, Sofia; Masquelier, Donald A.; Merrill, Landon; Terry, Jessica M.; Mudivarti, Patrice A.; Wyatt, Paul W.; Bharadwaj, Rajiv; Makarewicz, Anthony J.; Li, Yuan (March 2016). "Haplotyping germline and cancer genomes with high-throughput linked-read sequencing". Nature Biotechnology. 34 (3): 303–311. doi:10.1038/nbt.3432. ISSN 1546-1696. PMC 4786454. PMID 26829319.
- 1 2 3 4 5 6 Mostovoy, Yulia; Levy-Sakin, Michal; Lam, Jessica; Lam, Ernest T; Hastie, Alex R; Marks, Patrick; Lee, Joyce; Chu, Catherine; Lin, Chin; Džakula, Željko; Cao, Han; Schlebusch, Stephen A; Giorda, Kristina; Schnall-Levin, Michael; Wall, Jeffrey D (July 2016). "A hybrid approach for de novo human genome sequence assembly and phasing". Nature Methods. 13 (7): 587–590. doi:10.1038/nmeth.3865. ISSN 1548-7091. PMC 4927370. PMID 27159086.
- 1 2 3 Elyanow, Rebecca; Wu, Hsin-Ta; Raphael, Benjamin J (2018-01-15). Curtis, Christina (ed.). "Identifying structural variants using linked-read sequencing data". Bioinformatics. 34 (2): 353–360. doi:10.1093/bioinformatics/btx712. ISSN 1367-4803. PMC 5860216. PMID 29112732.
- 1 2 3 Ott, Alina; Schnable, James C.; Yeh, Cheng-Ting; Wu, Linjiang; Liu, Chao; Hu, Heng-Cheng; Dalgard, Clifton L.; Sarkar, Soumik; Schnable, Patrick S. (2018-09-04). "Linked read technology for assembling large complex and polyploid genomes". BMC Genomics. 19 (1): 651. doi:10.1186/s12864-018-5040-z. ISSN 1471-2164. PMC 6122573. PMID 30180802.
- 1 2 Martinez-Viaud, Karine A; Lawley, Cindy Taylor; Vergara, Milmer Martinez; Ben-Zvi, Gil; Biniashvili, Tammy; Baruch, Kobi; St. Leger, Judy; Le, Jennie; Natarajan, Aparna; Rivera, Marlem; Guillergan, Marbie; Jaeger, Erich; Steffy, Brian; Zimin, Aleksey (2019-03-01). "New de novo assembly of the Atlantic bottlenose dolphin ( Tursiops truncatus ) improves genome completeness and provides haplotype phasing". GigaScience. 8 (3). doi:10.1093/gigascience/giy168. ISSN 2047-217X. PMC 6443575. PMID 30698692.
- ↑ Wu, Shan; Lau, Kin H.; Cao, Qinghe; Hamilton, John P.; Sun, Honghe; Zhou, Chenxi; Eserman, Lauren; Gemenet, Dorcus C.; Olukolu, Bode A.; Wang, Haiyan; Crisovan, Emily; Godden, Grant T.; Jiao, Chen; Wang, Xin; Kitavi, Mercy (2018-11-02). "Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement". Nature Communications. 9 (1): 4580. Bibcode:2018NatCo...9.4580W. doi:10.1038/s41467-018-06983-8. ISSN 2041-1723. PMC 6214957. PMID 30389915.
- 1 2 3 Kronenberg, Zev N.; Fiddes, Ian T.; Gordon, David; Murali, Shwetha; Cantsilieris, Stuart; Meyerson, Olivia S.; Underwood, Jason G.; Nelson, Bradley J.; Chaisson, Mark J. P.; Dougherty, Max L.; Munson, Katherine M.; Hastie, Alex R.; Diekhans, Mark; Hormozdiari, Fereydoun; Lorusso, Nicola (2018-06-08). "High-resolution comparative analysis of great ape genomes". Science. 360 (6393): eaar6343. doi:10.1126/science.aar6343. ISSN 0036-8075. PMC 6178954. PMID 29880660.
- ↑ Maestri, Simone; Maturo, Maria Giovanna; Cosentino, Emanuela; Marcolungo, Luca; Iadarola, Barbara; Fortunati, Elisabetta; Rossato, Marzia; Delledonne, Massimo (2020-12-01). "A Long-Read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings". International Journal of Molecular Sciences. 21 (23): 9177. doi:10.3390/ijms21239177. ISSN 1422-0067. PMC 7731377. PMID 33271988.
- 1 2 3 Jang, Se Song; Lim, Byung Chan; Yoo, Seong-Keun; Shin, Jong-Yeon; Kim, Ki-Joong; Seo, Jeong-Sun; Kim, Jong-Il; Chae, Jong Hee (2018-06-06). "Targeted linked-read sequencing for direct haplotype phasing of maternal DMD alleles: a practical and reliable method for noninvasive prenatal diagnosis". Scientific Reports. 8 (1): 8678. Bibcode:2018NatSR...8.8678J. doi:10.1038/s41598-018-26941-0. ISSN 2045-2322. PMC 5989205. PMID 29875376.
- ↑ Ostendorf, Benjamin N.; Bilanovic, Jana; Adaku, Nneoma; Tafreshian, Kimia N.; Tavora, Bernardo; Vaughan, Roger D.; Tavazoie, Sohail F. (July 2020). "Common germline variants of the human APOE gene modulate melanoma progression and survival". Nature Medicine. 26 (7): 1048–1053. doi:10.1038/s41591-020-0879-3. ISSN 1546-170X. PMC 8058866. PMID 32451497.
- 1 2 Tilgner, Hagen; Jahanbani, Fereshteh; Gupta, Ishaan; Collier, Paul; Wei, Eric; Rasmussen, Morten; Snyder, Michael (February 2018). "Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome". Genome Research. 28 (2): 231–242. doi:10.1101/gr.230516.117. ISSN 1088-9051. PMC 5793787. PMID 29196558.
- 1 2 3 Konki, Mikko; Malonzo, Maia; Karlsson, Ida K.; Lindgren, Noora; Ghimire, Bishwa; Smolander, Johannes; Scheinin, Noora M.; Ollikainen, Miina; Laiho, Asta; Elo, Laura L.; Lönnberg, Tapio; Röyttä, Matias; Pedersen, Nancy L.; Kaprio, Jaakko; Lähdesmäki, Harri (December 2019). "Peripheral blood DNA methylation differences in twin pairs discordant for Alzheimer's disease". Clinical Epigenetics. 11 (1): 130. doi:10.1186/s13148-019-0729-7. ISSN 1868-7075. PMC 6721173. PMID 31477183.
- ↑ McGrath-Morrow, Sharon A.; Ndeh, Roland; Helmin, Kathryn A.; Khuder, Basil; Rothblum-Oviatt, Cynthia; Collaco, Joseph M.; Wright, Jennifer; Reyfman, Paul A.; Lederman, Howard M.; Singer, Benjamin D. (2020-05-04). "DNA methylation and gene expression signatures are associated with ataxia-telangiectasia phenotype". Scientific Reports. 10 (1): 7479. Bibcode:2020NatSR..10.7479M. doi:10.1038/s41598-020-64514-2. ISSN 2045-2322. PMC 7198504. PMID 32366930.
- 1 2 "United States Court of Appeals for the Federal Circuit - Bio-Rad Laboratories Inc. v. 10x Genomics" (PDF). Cases.Justia.com. 2020-08-03.