 | |
| Clinical data | |
|---|---|
| Pronunciation | /proʊˈkeɪnəmaɪd/ |
| Trade names | Pronestyl, Procan, Procanbid, others |
| AHFS/Drugs.com | Monograph |
| Routes of administration | IV, IM, by mouth |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 85% (by mouth) |
| Protein binding | 15 to 20% |
| Metabolism | Liver (CYP2D6-mediated) |
| Elimination half-life | ~2.5 to 4.5 hours |
| Excretion | Kidney |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.000.072 |
| Chemical and physical data | |
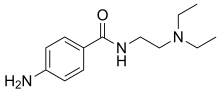
| Formula | C13H21N3O |
| Molar mass | 235.331 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| (verify) | |
Procainamide (PCA) is a medication of the antiarrhythmic class used for the treatment of cardiac arrhythmias. It is a sodium channel blocker of cardiomyocytes; thus it is classified by the Vaughan Williams classification system as class Ia. In addition to blocking the INa current, it inhibits the IKr rectifier K+ current.[1] Procainamide is also known to induce a voltage-dependent open channel block on the batrachotoxin (BTX)-activated sodium channels in cardiomyocytes.[2]
Uses
Medical
Procainamide is used for treating ventricular arrhythmias: ventricular ectopy and tachycardia and supraventricular arrhythmias: atrial fibrillation, and re-entrant and automatic supraventricular tachycardia.[3] For example, it can be used to convert new-onset atrial fibrillation, and although was initially thought to be suboptimal for this purpose, a growing body of literature is amounting in support for this exact cause.[4][5]
It is administered by mouth, by intramuscular injection, or intravenously.[6][7]
Others
It has also been used as a chromatography resin because it somewhat binds protein.[8][9][10][11]
Side effects
There are many side effects following the induction of procainamide. These adverse effects are ventricular dysrhythmia, bradycardia, hypotension and shock. The adverse effects occur even more often if the daily doses are increased. Procainamide may also lead to drug fever and other allergic responses. There is also a chance that drug-induced lupus erythematosus occurs, which at the same time leads to arthralgia, myalgia and pleurisy. Most of these side effects may occur due to the acetylation of procainamide.[12]
Toxicity
There is a close line between the plasma concentrations of the therapeutic and toxic effect, therefore a high risk for toxicity.[12] Many symptoms resemble systemic lupus erythematosus because procainamide reactivates hydroxylamine and nitroso metabolites, which bind to histone proteins and are toxic to lymphocytes. The hydroxylamine and nitroso metabolites are also toxic to bone marrow cells and can cause agranulocytosis. These metabolites are formed due to the activation of polymorphonuclear leukocytes. These leukocytes release myeloperoxidase and hydrogen peroxide, which oxidize the primary aromatic amine of procainamide to form procainamide hydroxylamine. The release of hydrogen peroxide is also called a respiratory burst, which occurs for procainamide in monocytes but not in lymphocytes. Furthermore, the metabolites can be formed by activated neutrophils. These metabolites could then bind to their cell membranes and cause a release of autoantibodies which would react with the neutrophils.[13] Procainamide hydroxylamine has more cytotoxicity by hindering the response of lymphocytes to T-cell and B-cell mitogens. Hydroxylamine can also generate methemoglobin, a protein that could hinder further oxygen exchange.[14]
It was also detected that the antiarrhythmic drug procainamide interferes with pacemakers. A toxic level of procainamide leads to decrease in ventricular conduction velocity and increase of the ventricular refractory period. This results in a disturbance in the artificial membrane potential and leads to a supraventricular tachycardia which induces failure of the pacemaker and death.[15] Thus, it prolongs QT interval of action potential and increases the risk of torsade de pointes.[1]
Procainamide could initiate leukopenia and/or agranulocytosis, which are serious hematologic disorders, and is also known for causing gastrointestinal disturbances and aggravating pre-existing abnormalities in impulse initiation and propagation.[3]
Pharmacology
Mechanism of action
Procainamide works as an anti-arrhythmic agent and is used to treat cardiac arrhythmia. It induces rapid block of the batrachotoxin (BTX)-activated sodium channels of the heart muscle and acts as antagonist to long-gating closures. The block is voltage-dependent and can occur from both sides; either from the intracellular or the extracellular side. Blocking from the extracellular side is weaker than from the intracellular side because it occurs via the hydrophobic pathway. Procainamide is present in charged form and probably requires a direct hydrophobic access to the binding site for blocking of the channel. Furthermore, blocking of the channel shows a decreased voltage sensitivity, which may result from the loss of voltage dependence of the blocking rate. Due to its charged and hydrophilic form, procainamide has its effect from the internal side, where it causes blockage of voltage-dependent, open channels. With increasing concentration of procainamide, the frequency of long blockage becomes less without the duration of blockage being affected. The rate of fast blocking is determined by the membrane depolarization. Membrane depolarization leads to increased blocking and decreased unblocking of the channels. Procainamide slows the conduction velocity and increases the refractory period, such that the maximal rate of depolarization is reduced.[2]
Metabolism
Procainamide is metabolized via different pathways. The most common one is the acetylation of procainamide to the less-toxic N-acetylprocainamide.[16] The rate of acetylation is genetically determined. There are two phenotypes that result from the acetylation process, namely the slow and rapid acetylator. Procainamide can also be oxidized by the cytochrome P-450 to a reactive oxide metabolite. But it seems that acetylation of the nitrogen group of procainamide decrease the amount of the chemical that would be available for the oxidative route.[17] Other metabolites of procainamide include desethyl-N-acetylprocainamide, desethylprocainamide, p-aminobenzoic acid, which are excreted via the urine. N-acetyl-4-aminobenzoic acid as well as N-acetyl-3-hydroxyprocainamide, N-acetylprocainamide-N-oxide and N-acetyl-4-aminohippuric acid are also metabolites of procainamide.[17]
Chemistry
4-amino-N-2-(diethylamino)ethyl-benzamide (also known as para-amino-N-2-(diethylamino)ethyl-benzamide because the amino substituent is attached to the para-position, Arene substitution patterns of the benzene ring) is a synthetic organic compound with the chemical formula C13-H21-N3-O.[18]
Procainamide is structurally similar to procaine, but in place of an ester group, procainamide contains an amide group. This substitution is the reason why procainamide exhibits a longer half-life time than procaine.[19][20]
Procainamide belongs to the aminobenzamides. These are aromatic carboxylic acid derivatives consisting of an amide with a benzamide moiety and a triethylamine attached to the amide nitrogen.[18][21][22]
In certain lines, the para-amino group might become a target site to attach further paraphernalia, e.g. ref. Ex18 in U.S. Patent 7,115,750.
History
Procainamide was approved by the US FDA on June 2, 1950, under the brand name "Pronestyl".[23] It was launched by Bristol-Myers Squibb in 1951.[24] Due to the loss of Indonesia in World War II, the source for cinchona alkaloids, a precursor of quinidine, was reduced. This led to research for a new antiarrhythmic drug. As a result, procaine was discovered, which has similar cardiac effects as quinidine.[25] In 1936 it was found by Mautz that by applying it directly on the myocardium, the ventricular threshold for electrical stimulation was elevated.[24] This mechanism is responsible for the antiarrhythmic effect. However, due to the short duration of action, caused by rapid enzymatic hydrolysis, its therapeutic applications were limited.[26] In addition, procaine also caused tremors and respiratory depression.[26][27] All these adverse features stimulated the search for an alternative to procaine. Studies were done on various congeners and metabolites and this ultimately led to the discovery of procainamide by Mark et al. It was found that procainamide was effective for treating ventricular arrhythmias, but it had the same toxicity profile as quinidine, and it could cause systemic lupus erythematosus-like syndrome.[25][27] These negative characteristics slowed the search for new antiarrhythmics based on the chemical structure of procainamide. In 1970 only five drugs were reported. These were the cardiac glycosides, quinidine, propranolol, lidocaine and diphenylhydantoin. In January 1996, extended release procainamide hydrochloride (Procanbid extended-release tablets) was approved by the FDA.[28]
References
- 1 2 Osadchii OE (August 2014). "Procainamide and lidocaine produce dissimilar changes in ventricular repolarization and arrhythmogenicity in guinea-pig". Fundamental & Clinical Pharmacology. 28 (4): 382–393. doi:10.1111/fcp.12046. PMID 23952942. S2CID 5086017.
- 1 2 Zamponi GW, Sui X, Codding PW, French RJ (December 1993). "Dual actions of procainamide on batrachotoxin-activated sodium channels: open channel block and prevention of inactivation". Biophysical Journal. 65 (6): 2324–2334. Bibcode:1993BpJ....65.2324Z. doi:10.1016/S0006-3495(93)81291-8. PMC 1225974. PMID 8312472.
- 1 2 Gould LA, ed. (1983). Drug Treatment of Cardiac Arrhythmias. Mount Kisco: Futura Publishing Company. pp. 73–74. ISBN 0879931906.
{{cite book}}: CS1 maint: overridden setting (link) - ↑ Stiell IG, Sivilotti ML, Taljaard M, Birnie D, Vadeboncoeur A, Hohl CM, et al. (February 2020). "Electrical versus pharmacological cardioversion for emergency department patients with acute atrial fibrillation (RAFF2): a partial factorial randomised trial". Lancet. 395 (10221): 339–349. doi:10.1016/S0140-6736(19)32994-0. PMID 32007169. S2CID 210978499.
- ↑ Fenster PE, Comess KA, Marsh R, Katzenberg C, Hager WD (September 1983). "Conversion of atrial fibrillation to sinus rhythm by acute intravenous procainamide infusion". American Heart Journal. 106 (3): 501–504. doi:10.1016/0002-8703(83)90692-0. PMID 6881022.
- ↑ Koch-Weser J, Klein SW (March 1971). "Procainamide dosage schedules, plasma concentrations, and clinical effects". JAMA. 215 (9): 1454–1460. doi:10.1001/jama.1971.03180220036006. PMID 5107621.
- ↑ Antman EM, Sabatine MS, eds. (2013). Cardiovascular Therapeutics: A companion to Braunwald's heart disease (4th ed.). Philadelphia, PA: Elsevier/Saunders. p. 410. ISBN 978-1-4557-0101-8.
{{cite book}}: CS1 maint: overridden setting (link) - ↑ "Procainamide Sepharose 4 Fast Flow". GE Healthcare Life Sciences. Archived from the original on 2021-08-29. Retrieved 2017-07-24.
- ↑ De la Hoz D, Doctor BP, Ralston JS, Rush RS, Wolfe AD (July 1986). "A simplified procedure for the purification of large quantities of fetal bovine serum acetylcholinesterase". Life Sciences. 39 (3): 195–199. doi:10.1016/0024-3205(86)90530-8. PMID 3736320.
- ↑ Ralston JS, Main AR, Kilpatrick BF, Chasson AL (April 1983). "Use of procainamide gels in the purification of human and horse serum cholinesterases". The Biochemical Journal. 211 (1): 243–250. doi:10.1042/bj2110243. PMC 1154348. PMID 6870822.
- ↑ Saxena A, Luo C, Doctor BP (October 2008). "Developing procedures for the large-scale purification of human serum butyrylcholinesterase". Protein Expression and Purification. 61 (2): 191–196. doi:10.1016/j.pep.2008.05.021. PMID 18602477.
- 1 2 Lawson DH, Jick H (October 1977). "Adverse reactions to procainamide". British Journal of Clinical Pharmacology. 4 (5): 507–511. doi:10.1111/j.1365-2125.1977.tb00777.x. PMC 1429167. PMID 911600.
- ↑ Uetrecht J, Zahid N, Rubin R (January 1988). "Metabolism of procainamide to a hydroxylamine by human neutrophils and mononuclear leukocytes". Chemical Research in Toxicology. 1 (1): 74–78. doi:10.1021/tx00001a013. PMID 2979715.
- ↑ Roberts SM, Adams LE, Donovan-Brand R, Budinsky R, Skoulis NP, Zimmer H, Hess EV (1989). "Procainamide hydroxylamine lymphocyte toxicity--I. Evidence for participation by hemoglobin". International Journal of Immunopharmacology. 11 (4): 419–427. doi:10.1016/0192-0561(89)90089-1. PMID 2476407.
- ↑ Gay RJ, Brown DF (November 1974). "Pacemaker failure due to procainamide toxicity". The American Journal of Cardiology. 34 (6): 728–732. doi:10.1016/0002-9149(74)90164-7. PMID 4422040.
- ↑ Roden DM, Reele SB, Higgins SB, Wilkinson GR, Smith RF, Oates JA, Woosley RL (September 1980). "Antiarrhythmic efficacy, pharmacokinetics and safety of N-acetylprocainamide in human subjects: comparison with procainamide". The American Journal of Cardiology. 46 (3): 463–468. doi:10.1016/0002-9149(80)90016-8. PMID 6158263.
- 1 2 Uetrecht JP, Freeman RW, Woosley RL (August 1981). "The implications of procainamide metabolism to its induction of lupus". Arthritis and Rheumatism. 24 (8): 994–1003. doi:10.1002/art.1780240803. PMID 6169352.
- 1 2 "Procainamide". www.drugbank.ca. 27 June 2018. Retrieved 28 June 2018.
- ↑ Adams HR (1995). Drugs Acting on the Cardiovascular System. Veterinary Pharmacology and Therapeutics (7th ed.). pp. 451–500.
- ↑ Plumb DC (1999). Veterinary Drug Handbook. White Bear Lake, USA: PharmaVet Publishing.
- ↑ EBI Web Team. "CHEBI:8428 - procainamide". www.ebi.ac.uk. Retrieved 28 June 2018.
- ↑ DeRuiter J (2005). "Amides and Related Functional Groups". Principles of Drug Action. p. 1.
- ↑ US Food and Drug Administration. "Drugs at FDA: FDA Approved Drug Products". USA: U.S. Food and Drug Administration (FDA). Retrieved 2012-08-13.
- 1 2 Hollman A (February 1992). "Procaine and procainamide". British Heart Journal. 67 (2): 143. doi:10.1136/hrt.67.2.143. PMC 1024743. PMID 18610401.
- 1 2 Walker MJ (January 2006). "Antiarrhythmic drug research". British Journal of Pharmacology. 147 (Suppl 1): S222–S231. doi:10.1038/sj.bjp.0706500. PMC 1760742. PMID 16402108.
- 1 2 Moe GK, Abildskov A (1965). "Antiarrhythmic drugs". In Goodman LS, Gilman A (eds.). Goodman and Gilman's The Pharmacological Basis of Therapeutics (3rd ed.). New York: Macmillan. pp. 699–715.
- 1 2 Lüderitz BB, ed. (2002). "Historical development of antiarrhythmic drug therapy". History of Disorders of Cardiac Rhythm (3rd ed.). New York: Wiley-Blackwell. pp. 87–114.
- ↑ Mishina E, Marroum P (2002). "Center for Drug Evaluation and Research Approval Package For: Application Number NDA 20-545/S007" (PDF). Clinical Pharmacology and Bioharmaceutics Review.