| Nobelium | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation |
| |||||||||||||||||||||||||||||||||||||||||||||||
| Mass number | [259] | |||||||||||||||||||||||||||||||||||||||||||||||
| Nobelium in the periodic table | ||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 102 | |||||||||||||||||||||||||||||||||||||||||||||||
| Group | f-block groups (no number) | |||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 7 | |||||||||||||||||||||||||||||||||||||||||||||||
| Block | f-block | |||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Rn] 5f14 7s2 | |||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 18, 32, 32, 8, 2 | |||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | ||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid (predicted)[1] | |||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | 1100 K (800 °C, 1500 °F) (predicted)[1] | |||||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 9.9(4) g/cm3 (predicted)[2][lower-alpha 1] | |||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | ||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | +2, +3 | |||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.3 (predicted)[3] | |||||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
| |||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | ||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | synthetic | |||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | face-centered cubic (fcc) (predicted)[2] | |||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 10028-14-5 | |||||||||||||||||||||||||||||||||||||||||||||||
| History | ||||||||||||||||||||||||||||||||||||||||||||||||
| Naming | after Alfred Nobel | |||||||||||||||||||||||||||||||||||||||||||||||
| Discovery | Joint Institute for Nuclear Research (1966) | |||||||||||||||||||||||||||||||||||||||||||||||
| Isotopes of nobelium | ||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||
Nobelium is a synthetic chemical element; it has symbol No and atomic number 102. It is named in honor of Alfred Nobel, the inventor of dynamite and benefactor of science. A radioactive metal, it is the tenth transuranic element and is the penultimate member of the actinide series. Like all elements with atomic number over 100, nobelium can only be produced in particle accelerators by bombarding lighter elements with charged particles. A total of twelve nobelium isotopes are known to exist; the most stable is 259No with a half-life of 58 minutes, but the shorter-lived 255No (half-life 3.1 minutes) is most commonly used in chemistry because it can be produced on a larger scale.
Chemistry experiments have confirmed that nobelium behaves as a heavier homolog to ytterbium in the periodic table. The chemical properties of nobelium are not completely known: they are mostly only known in aqueous solution. Before nobelium's discovery, it was predicted that it would show a stable +2 oxidation state as well as the +3 state characteristic of the other actinides; these predictions were later confirmed, as the +2 state is much more stable than the +3 state in aqueous solution and it is difficult to keep nobelium in the +3 state.
In the 1950s and 1960s, many claims of the discovery of nobelium were made from laboratories in Sweden, the Soviet Union, and the United States. Although the Swedish scientists soon retracted their claims, the priority of the discovery and therefore the naming of the element was disputed between Soviet and American scientists. It was not until 1997 that the International Union of Pure and Applied Chemistry (IUPAC) credited the Soviet team with the discovery. Even so, nobelium, the Swedish proposal, was retained as the name of the element due to its long-standing use in the literature.
Introduction
Synthesis of superheavy nuclei

A superheavy[lower-alpha 2] atomic nucleus is created in a nuclear reaction that combines two other nuclei of unequal size[lower-alpha 3] into one; roughly, the more unequal the two nuclei in terms of mass, the greater the possibility that the two react.[11] The material made of the heavier nuclei is made into a target, which is then bombarded by the beam of lighter nuclei. Two nuclei can only fuse into one if they approach each other closely enough; normally, nuclei (all positively charged) repel each other due to electrostatic repulsion. The strong interaction can overcome this repulsion but only within a very short distance from a nucleus; beam nuclei are thus greatly accelerated in order to make such repulsion insignificant compared to the velocity of the beam nucleus.[12] The energy applied to the beam nuclei to accelerate them can cause them to reach speeds as high as one-tenth of the speed of light. However, if too much energy is applied, the beam nucleus can fall apart.[12]
Coming close enough alone is not enough for two nuclei to fuse: when two nuclei approach each other, they usually remain together for approximately 10−20 seconds and then part ways (not necessarily in the same composition as before the reaction) rather than form a single nucleus.[12][13] This happens because during the attempted formation of a single nucleus, electrostatic repulsion tears apart the nucleus that is being formed.[12] Each pair of a target and a beam is characterized by its cross section—the probability that fusion will occur if two nuclei approach one another expressed in terms of the transverse area that the incident particle must hit in order for the fusion to occur.[lower-alpha 4] This fusion may occur as a result of the quantum effect in which nuclei can tunnel through electrostatic repulsion. If the two nuclei can stay close for past that phase, multiple nuclear interactions result in redistribution of energy and an energy equilibrium.[12]
| External videos | |
|---|---|
The resulting merger is an excited state[16]—termed a compound nucleus—and thus it is very unstable.[12] To reach a more stable state, the temporary merger may fission without formation of a more stable nucleus.[17] Alternatively, the compound nucleus may eject a few neutrons, which would carry away the excitation energy; if the latter is not sufficient for a neutron expulsion, the merger would produce a gamma ray. This happens in approximately 10−16 seconds after the initial nuclear collision and results in creation of a more stable nucleus.[17] The definition by the IUPAC/IUPAP Joint Working Party (JWP) states that a chemical element can only be recognized as discovered if a nucleus of it has not decayed within 10−14 seconds. This value was chosen as an estimate of how long it takes a nucleus to acquire its outer electrons and thus display its chemical properties.[18][lower-alpha 5]
Decay and detection
The beam passes through the target and reaches the next chamber, the separator; if a new nucleus is produced, it is carried with this beam.[20] In the separator, the newly produced nucleus is separated from other nuclides (that of the original beam and any other reaction products)[lower-alpha 6] and transferred to a surface-barrier detector, which stops the nucleus. The exact location of the upcoming impact on the detector is marked; also marked are its energy and the time of the arrival.[20] The transfer takes about 10−6 seconds; in order to be detected, the nucleus must survive this long.[23] The nucleus is recorded again once its decay is registered, and the location, the energy, and the time of the decay are measured.[20]
Stability of a nucleus is provided by the strong interaction. However, its range is very short; as nuclei become larger, its influence on the outermost nucleons (protons and neutrons) weakens. At the same time, the nucleus is torn apart by electrostatic repulsion between protons, and its range is not limited.[24] Total binding energy provided by the strong interaction increases linearly with the number of nucleons, whereas electrostatic repulsion increases with the square of the atomic number, i.e. the latter grows faster and becomes increasingly important for heavy and superheavy nuclei.[25][26] Superheavy nuclei are thus theoretically predicted[27] and have so far been observed[28] to predominantly decay via decay modes that are caused by such repulsion: alpha decay and spontaneous fission.[lower-alpha 7] Almost all alpha emitters have over 210 nucleons,[30] and the lightest nuclide primarily undergoing spontaneous fission has 238.[31] In both decay modes, nuclei are inhibited from decaying by corresponding energy barriers for each mode, but they can be tunnelled through.[25][26]

Alpha particles are commonly produced in radioactive decays because mass of an alpha particle per nucleon is small enough to leave some energy for the alpha particle to be used as kinetic energy to leave the nucleus.[33] Spontaneous fission is caused by electrostatic repulsion tearing the nucleus apart and produces various nuclei in different instances of identical nuclei fissioning.[26] As the atomic number increases, spontaneous fission rapidly becomes more important: spontaneous fission partial half-lives decrease by 23 orders of magnitude from uranium (element 92) to nobelium (element 102),[34] and by 30 orders of magnitude from thorium (element 90) to fermium (element 100).[35] The earlier liquid drop model thus suggested that spontaneous fission would occur nearly instantly due to disappearance of the fission barrier for nuclei with about 280 nucleons.[26][36] The later nuclear shell model suggested that nuclei with about 300 nucleons would form an island of stability in which nuclei will be more resistant to spontaneous fission and will primarily undergo alpha decay with longer half-lives.[26][36] Subsequent discoveries suggested that the predicted island might be further than originally anticipated; they also showed that nuclei intermediate between the long-lived actinides and the predicted island are deformed, and gain additional stability from shell effects.[37] Experiments on lighter superheavy nuclei,[38] as well as those closer to the expected island,[34] have shown greater than previously anticipated stability against spontaneous fission, showing the importance of shell effects on nuclei.[lower-alpha 8]
Alpha decays are registered by the emitted alpha particles, and the decay products are easy to determine before the actual decay; if such a decay or a series of consecutive decays produces a known nucleus, the original product of a reaction can be easily determined.[lower-alpha 9] (That all decays within a decay chain were indeed related to each other is established by the location of these decays, which must be in the same place.)[20] The known nucleus can be recognized by the specific characteristics of decay it undergoes such as decay energy (or more specifically, the kinetic energy of the emitted particle).[lower-alpha 10] Spontaneous fission, however, produces various nuclei as products, so the original nuclide cannot be determined from its daughters.[lower-alpha 11]
The information available to physicists aiming to synthesize a superheavy element is thus the information collected at the detectors: location, energy, and time of arrival of a particle to the detector, and those of its decay. The physicists analyze this data and seek to conclude that it was indeed caused by a new element and could not have been caused by a different nuclide than the one claimed. Often, provided data is insufficient for a conclusion that a new element was definitely created and there is no other explanation for the observed effects; errors in interpreting data have been made.[lower-alpha 12]Discovery

The discovery of element 102 was a complicated process and was claimed by groups from Sweden, the United States, and the Soviet Union. The first complete and incontrovertible report of its detection only came in 1966 from the Joint Institute of Nuclear Research at Dubna (then in the Soviet Union).[49]
The first announcement of the discovery of element 102 was announced by physicists at the Nobel Institute in Sweden in 1957. The team reported that they had bombarded a curium target with carbon-13 ions for twenty-five hours in half-hour intervals. Between bombardments, ion-exchange chemistry was performed on the target. Twelve out of the fifty bombardments contained samples emitting (8.5 ± 0.1) MeV alpha particles, which were in drops which eluted earlier than fermium (atomic number Z = 100) and californium (Z = 98). The half-life reported was 10 minutes and was assigned to either 251102 or 253102, although the possibility that the alpha particles observed were from a presumably short-lived mendelevium (Z = 101) isotope created from the electron capture of element 102 was not excluded.[49] The team proposed the name nobelium (No) for the new element,[50][51] which was immediately approved by IUPAC,[52] a decision which the Dubna group characterized in 1968 as being hasty.[53]
In 1958, scientists at the Lawrence Berkeley National Laboratory repeated the experiment. The Berkeley team, consisting of Albert Ghiorso, Glenn T. Seaborg, John R. Walton and Torbjørn Sikkeland, used the new heavy-ion linear accelerator (HILAC) to bombard a curium target (95% 244Cm and 5% 246Cm) with 13C and 12C ions. They were unable to confirm the 8.5 MeV activity claimed by the Swedes but were instead able to detect decays from fermium-250, supposedly the daughter of 254102 (produced from the curium-246), which had an apparent half-life of ~3 s.[49]
In 1959, the Swedish team attempted to explain the Berkeley team's inability to detect element 102 in 1958, maintaining that they did discover it. However, later work has shown that no nobelium isotopes lighter than 259No (no heavier isotopes could have been produced in the Swedish experiments) with a half-life over 3 minutes exist, and that the Swedish team's results are most likely from thorium-225, which has a half-life of 8 minutes and quickly undergoes triple alpha decay to polonium-213, which has a decay energy of 8.53612 MeV. This hypothesis is lent weight by the fact that thorium-225 can easily be produced in the reaction used and would not be separated out by the chemical methods used. Later work on nobelium also showed that the divalent state is more stable than the trivalent one and hence that the samples emitting the alpha particles could not have contained nobelium, as the divalent nobelium would not have eluted with the other trivalent actinides.[49] Thus, the Swedish team later retracted their claim and associated the activity to background effects.[52]
Later 1963 Dubna work confirmed that 254102 could be produced in this reaction, but that its half-life was actually 50±10 s. In 1967, the Berkeley team attempted to defend their work, stating that the isotope found was indeed 250Fm but the isotope that the half-life measurements actually related to was californium-244, granddaughter of 252102, produced from the more abundant curium-244. Energy differences were then attributed to "resolution and drift problems", although these had not been previously reported and should also have influenced other results. 1977 experiments showed that 252102 indeed had a 2.3-second half-life. However, 1973 work also showed that the 250Fm recoil could have also easily been produced from the isomeric transition of 250mFm (half-life 1.8 s) which could also have been formed in the reaction at the energy used.[49] Given this, it is probable that no nobelium was actually produced in this experiment.[49]
In 1959, the team continued their studies and claimed that they were able to produce an isotope that decayed predominantly by emission of an 8.3 MeV alpha particle, with a half-life of 3 s with an associated 30% spontaneous fission branch. The activity was initially assigned to 254102 but later changed to 252102. However, they also noted that it was not certain that nobelium had been produced due to difficult conditions.[49] The Berkeley team decided to adopt the proposed name of the Swedish team, "nobelium", for the element.[52]
- 244
96Cm
+ 12
6C
→ 256
102No
*
→ 252
102No
+ 4 1
0
n
Meanwhile, in Dubna, experiments were carried out in 1958 and 1960 aiming to synthesize element 102 as well. The first 1958 experiment bombarded plutonium-239 and -241 with oxygen-16 ions. Some alpha decays with energies just over 8.5 MeV were observed, and they were assigned to 251,252,253102, although the team wrote that formation of isotopes from lead or bismuth impurities (which would not produce nobelium) could not be ruled out. While later 1958 experiments noted that new isotopes could be produced from mercury, thallium, lead, or bismuth impurities, the scientists still stood by their conclusion that element 102 could be produced from this reaction, mentioning a half-life of under 30 seconds and a decay energy of (8.8 ± 0.5) MeV. Later 1960 experiments proved that these were background effects. 1967 experiments also lowered the decay energy to (8.6 ± 0.4) MeV, but both values are too high to possibly match those of 253No or 254No.[49] The Dubna team later stated in 1970 and again in 1987 that these results were not conclusive.[49]
In 1961, Berkeley scientists claimed the discovery of element 103 in the reaction of californium with boron and carbon ions. They claimed the production of the isotope 257103, and also claimed to have synthesized an alpha decaying isotope of element 102 that had a half-life of 15 s and alpha decay energy 8.2 MeV. They assigned this to 255102 without giving a reason for the assignment. The values do not agree with those now known for 255No, although they do agree with those now known for 257No, and while this isotope probably played a part in this experiment, its discovery was inconclusive.[49]
Work on element 102 also continued in Dubna, and in 1964, experiments were carried out there to detect alpha-decay daughters of element 102 isotopes by synthesizing element 102 from the reaction of a uranium-238 target with neon ions. The products were carried along a silver catcher foil and purified chemically, and the isotopes 250Fm and 252Fm were detected. The yield of 252Fm was interpreted as evidence that its parent 256102 was also synthesized: as it was noted that 252Fm could also be produced directly in this reaction by the simultaneous emission of an alpha particle with the excess neutrons, steps were taken to ensure that 252Fm could not go directly to the catcher foil. The half-life detected for 256102 was 8 s, which is much higher than the more modern 1967 value of (3.2 ± 0.2) s.[49] Further experiments were conducted in 1966 for 254102, using the reactions 243Am(15N,4n)254102 and 238U(22Ne,6n)254102, finding a half-life of (50 ± 10) s: at that time the discrepancy between this value and the earlier Berkeley value was not understood, although later work proved that the formation of the isomer 250mFm was less likely in the Dubna experiments than at the Berkeley ones. In hindsight, the Dubna results on 254102 were probably correct and can be now considered a conclusive detection of element 102.[49]
One more very convincing experiment from Dubna was published in 1966, again using the same two reactions, which concluded that 254102 indeed had a half-life much longer than the 3 seconds claimed by Berkeley.[49] Later work in 1967 at Berkeley and 1971 at the Oak Ridge National Laboratory fully confirmed the discovery of element 102 and clarified earlier observations.[52] In December 1966, the Berkeley group repeated the Dubna experiments and fully confirmed them, and used this data to finally assign correctly the isotopes they had previously synthesized but could not yet identify at the time, and thus claimed to have discovered nobelium in 1958 to 1961.[52]
- 238
92U
+ 22
10Ne
→ 260
102No
*
→ 254
102No
+ 6 1
0
n
In 1969, the Dubna team carried out chemical experiments on element 102 and concluded that it behaved as the heavier homologue of ytterbium. The Russian scientists proposed the name joliotium (Jo) for the new element after Irène Joliot-Curie, who had recently died, creating an element naming controversy that would not be resolved for several decades, with each group using its own proposed names.[52][54]
In 1992, the IUPAC-IUPAP Transfermium Working Group (TWG) reassessed the claims of discovery and concluded that only the Dubna work from 1966 correctly detected and assigned decays to nuclei with atomic number 102 at the time. The Dubna team are therefore officially recognised as the discoverers of nobelium although it is possible that it was detected at Berkeley in 1959.[49] This decision was criticized by Berkeley the following year, calling the reopening of the cases of elements 101 to 103 a "futile waste of time", while Dubna agreed with IUPAC's decision.[53]
In 1994, as part of an attempted resolution to the element naming controversy, IUPAC ratified names for elements 101–109. For element 102, it ratified the name nobelium (No) on the basis that it had become entrenched in the literature over the course of 30 years and that Alfred Nobel should be commemorated in this fashion.[55] Because of outcry over the 1994 names, which mostly did not respect the choices of the discoverers, a comment period ensued, and in 1995 IUPAC named element 102 flerovium (Fl) as part of a new proposal, after either Georgy Flyorov or his eponymous Flerov Laboratory of Nuclear Reactions.[56] This proposal was also not accepted, and in 1997 the name nobelium was restored.[55] Today the name flerovium, with the same symbol, refers to element 114.[57]
Characteristics
Physical
In the periodic table, nobelium is located to the right of the actinide mendelevium, to the left of the actinide lawrencium, and below the lanthanide ytterbium. Nobelium metal has not yet been prepared in bulk quantities, and bulk preparation is currently impossible.[59] Nevertheless, a number of predictions and some preliminary experimental results have been done regarding its properties.[59]
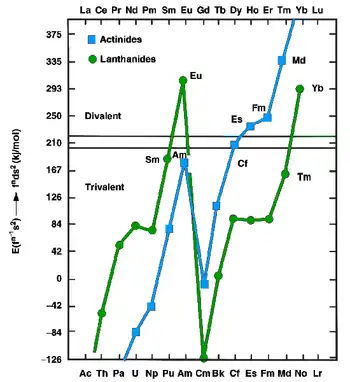
The lanthanides and actinides, in the metallic state, can exist as either divalent (such as europium and ytterbium) or trivalent (most other lanthanides) metals. The former have fns2 configurations, whereas the latter have fn−1d1s2 configurations. In 1975, Johansson and Rosengren examined the measured and predicted values for the cohesive energies (enthalpies of crystallization) of the metallic lanthanides and actinides, both as divalent and trivalent metals.[60][61] The conclusion was that the increased binding energy of the [Rn]5f136d17s2 configuration over the [Rn]5f147s2 configuration for nobelium was not enough to compensate for the energy needed to promote one 5f electron to 6d, as is true also for the very late actinides: thus einsteinium, fermium, mendelevium, and nobelium were expected to be divalent metals, although for nobelium this prediction has not yet been confirmed.[60] The increasing predominance of the divalent state well before the actinide series concludes is attributed to the relativistic stabilization of the 5f electrons, which increases with increasing atomic number: an effect of this is that nobelium is predominantly divalent instead of trivalent, unlike all the other lanthanides and actinides.[62] In 1986, nobelium metal was estimated to have an enthalpy of sublimation between 126 kJ/mol, a value close to the values for einsteinium, fermium, and mendelevium and supporting the theory that nobelium would form a divalent metal.[59] Like the other divalent late actinides (except the once again trivalent lawrencium), metallic nobelium should assume a face-centered cubic crystal structure.[2] Divalent nobelium metal should have a metallic radius of around 197 pm.[59] Nobelium's melting point has been predicted to be 800 °C, the same value as that estimated for the neighboring element mendelevium.[63] Its density is predicted to be around 9.9 ± 0.4 g/cm3.[2]
Chemical
The chemistry of nobelium is incompletely characterized and is known only in aqueous solution, in which it can take on the +3 or +2 oxidation states, the latter being more stable.[50] It was largely expected before the discovery of nobelium that in solution, it would behave like the other actinides, with the trivalent state being predominant; however, Seaborg predicted in 1949 that the +2 state would also be relatively stable for nobelium, as the No2+ ion would have the ground-state electron configuration [Rn]5f14, including the stable filled 5f14 shell. It took nineteen years before this prediction was confirmed.[64]
In 1967, experiments were conducted to compare nobelium's chemical behavior to that of terbium, californium, and fermium. All four elements were reacted with chlorine and the resulting chlorides were deposited along a tube, along which they were carried by a gas. It was found that the nobelium chloride produced was strongly adsorbed on solid surfaces, proving that it was not very volatile, like the chlorides of the other three investigated elements. However, both NoCl2 and NoCl3 were expected to exhibit nonvolatile behavior and hence this experiment was inconclusive as to what the preferred oxidation state of nobelium was.[64] Determination of nobelium's favoring of the +2 state had to wait until the next year, when cation-exchange chromatography and coprecipitation experiments were carried out on around fifty thousand 255No atoms, finding that it behaved differently from the other actinides and more like the divalent alkaline earth metals. This proved that in aqueous solution, nobelium is most stable in the divalent state when strong oxidizers are absent.[64] Later experimentation in 1974 showed that nobelium eluted with the alkaline earth metals, between Ca2+ and Sr2+.[64] Nobelium is the only known f-block element for which the +2 state is the most common and stable one in aqueous solution. This occurs because of the large energy gap between the 5f and 6d orbitals at the end of the actinide series.[65]
It is expected that the relativistic stabilization of the 7s subshell greatly destabilizes nobelium dihydride, NoH2, and relativistic stabilisation of the 7p1/2 spinor over the 6d3/2 spinor mean that excited states in nobelium atoms have 7s and 7p contribution instead of the expected 6d contribution. The long No–H distances in the NoH2 molecule and the significant charge transfer lead to extreme ionicity with a dipole moment of 5.94 D for this molecule. In this molecule, nobelium is expected to exhibit main-group-like behavior, specifically acting like an alkaline earth metal with its ns2 valence shell configuration and core-like 5f orbitals.[66]
Nobelium's complexing ability with chloride ions is most similar to that of barium, which complexes rather weakly.[64] Its complexing ability with citrate, oxalate, and acetate in an aqueous solution of 0.5 M ammonium nitrate is between that of calcium and strontium, although it is somewhat closer to that of strontium.[64]
The standard reduction potential of the E°(No3+→No2+) couple was estimated in 1967 to be between +1.4 and +1.5 V;[64] it was later found in 2009 to be only about +0.75 V.[67] The positive value shows that No2+ is more stable than No3+ and that No3+ is a good oxidizing agent. While the quoted values for the E°(No2+→No0) and E°(No3+→No0) vary among sources, the accepted standard estimates are −2.61 and −1.26 V.[64] It has been predicted that the value for the E°(No4+→No3+) couple would be +6.5 V.[64] The Gibbs energies of formation for No3+ and No2+ are estimated to be −342 and −480 kJ/mol, respectively.[64]
Atomic
A nobelium atom has 102 electrons. They are expected to be arranged in the configuration [Rn]5f147s2 (ground state term symbol 1S0), although experimental verification of this electron configuration had not yet been made as of 2006. The sixteen electrons in the 5f and 7s subshells are valence electrons.[59] In forming compounds, three valence electrons may be lost, leaving behind a [Rn]5f13 core: this conforms to the trend set by the other actinides with their [Rn]5fn electron configurations in the tripositive state. Nevertheless, it is more likely that only two valence electrons are lost, leaving behind a stable [Rn]5f14 core with a filled 5f14 shell. The first ionization potential of nobelium was measured to be at most (6.65 ± 0.07) eV in 1974, based on the assumption that the 7s electrons would ionize before the 5f ones;[68] this value has not yet been refined further due to nobelium's scarcity and high radioactivity.[69] The ionic radius of hexacoordinate and octacoordinate No3+ had been preliminarily estimated in 1978 to be around 90 and 102 pm respectively;[64] the ionic radius of No2+ has been experimentally found to be 100 pm to two significant figures.[59] The enthalpy of hydration of No2+ has been calculated as 1486 kJ/mol.[64]
Isotopes
Thirteen isotopes of nobelium are known, with mass numbers 249–260 and 262; all are radioactive.[70][71] Additionally, nuclear isomers are known for mass numbers 250, 251, 253, and 254.[72][73] Of these, the longest-lived isotope is 259No with a half-life of 58 minutes, and the longest-lived isomer is 251mNo with a half-life of 1.7 seconds.[72][73] However, the still undiscovered isotope 261No is predicted to have a still longer half-life of 170 min.[72][73] Additionally, the shorter-lived 255No (half-life 3.1 minutes) is more often used in chemical experimentation because it can be produced in larger quantities from irradiation of californium-249 with carbon-12 ions.[70] After 259No and 255No, the next most stable nobelium isotopes are 253No (half-life 1.62 minutes), 254No (51 seconds), 257No (25 seconds), 256No (2.91 seconds), and 252No (2.57 seconds).[70][72][73] All of the remaining nobelium isotopes have half-lives that are less than a second, and the shortest-lived known nobelium isotope (250No) has a half-life of only 0.25 milliseconds.[70][72][73] The isotope 254No is especially interesting theoretically as it is in the middle of a series of prolate nuclei from 231Pa to 279Rg, and the formation of its nuclear isomers (of which two are known) is controlled by proton orbitals such as 2f5/2 which come just above the spherical proton shell; it can be synthesized in the reaction of 208Pb with 48Ca.[74]
The half-lives of nobelium isotopes increase smoothly from 250No to 253No. However, a dip appears at 254No, and beyond this the half-lives of even-even nobelium isotopes drop sharply as spontaneous fission becomes the dominant decay mode. For example, the half-life of 256No is almost three seconds, but that of 258No is only 1.2 milliseconds.[70][72][73] This shows that at nobelium, the mutual repulsion of protons poses a limit to the region of long-lived nuclei in the actinide series.[75] The even-odd nobelium isotopes mostly continue to have longer half-lives as their mass numbers increase, with a dip in the trend at 257No.[70][72][73]
Preparation and purification
The isotopes of nobelium are mostly produced by bombarding actinide targets (uranium, plutonium, curium, californium, or einsteinium), with the exception of nobelium-262, which is produced as the daughter of lawrencium-262.[70] The most commonly used isotope, 255No, can be produced from bombarding curium-248 or californium-249 with carbon-12: the latter method is more common. Irradiating a 350 μg cm−2 target of californium-249 with three trillion (3 × 1012) 73 MeV carbon-12 ions per second for ten minutes can produce around 1200 nobelium-255 atoms.[70]
Once the nobelium-255 is produced, it can be separated out similarly as used to purify the neighboring actinide mendelevium. The recoil momentum of the produced nobelium-255 atoms is used to bring them physically far away from the target from which they are produced, bringing them onto a thin foil of metal (usually beryllium, aluminium, platinum, or gold) just behind the target in a vacuum: this is usually combined by trapping the nobelium atoms in a gas atmosphere (frequently helium), and carrying them along with a gas jet from a small opening in the reaction chamber. Using a long capillary tube, and including potassium chloride aerosols in the helium gas, the nobelium atoms can be transported over tens of meters.[76] The thin layer of nobelium collected on the foil can then be removed with dilute acid without completely dissolving the foil.[76] The nobelium can then be isolated by exploiting its tendency to form the divalent state, unlike the other trivalent actinides: under typically used elution conditions (bis-(2-ethylhexyl) phosphoric acid (HDEHP) as stationary organic phase and 0.05 M hydrochloric acid as mobile aqueous phase, or using 3 M hydrochloric acid as an eluant from cation-exchange resin columns), nobelium will pass through the column and elute while the other trivalent actinides remain on the column.[76] However, if a direct "catcher" gold foil is used, the process is complicated by the need to separate out the gold using anion-exchange chromatography before isolating the nobelium by elution from chromatographic extraction columns using HDEHP.[76]
Notes
- ↑ The density is calculated from the predicted metallic radius (Silva 2008, p. 1639) and the predicted close-packed crystal structure (Fournier 1976).
- ↑ In nuclear physics, an element is called heavy if its atomic number is high; lead (element 82) is one example of such a heavy element. The term "superheavy elements" typically refers to elements with atomic number greater than 103 (although there are other definitions, such as atomic number greater than 100[6] or 112;[7] sometimes, the term is presented an equivalent to the term "transactinide", which puts an upper limit before the beginning of the hypothetical superactinide series).[8] Terms "heavy isotopes" (of a given element) and "heavy nuclei" mean what could be understood in the common language—isotopes of high mass (for the given element) and nuclei of high mass, respectively.
- ↑ In 2009, a team at the JINR led by Oganessian published results of their attempt to create hassium in a symmetric 136Xe + 136Xe reaction. They failed to observe a single atom in such a reaction, putting the upper limit on the cross section, the measure of probability of a nuclear reaction, as 2.5 pb.[9] In comparison, the reaction that resulted in hassium discovery, 208Pb + 58Fe, had a cross section of ~20 pb (more specifically, 19+19
-11 pb), as estimated by the discoverers.[10] - ↑ The amount of energy applied to the beam particle to accelerate it can also influence the value of cross section. For example, in the 28
14Si
+ 1
0n
→ 28
13Al
+ 1
1p
reaction, cross section changes smoothly from 370 mb at 12.3 MeV to 160 mb at 18.3 MeV, with a broad peak at 13.5 MeV with the maximum value of 380 mb.[14] - ↑ This figure also marks the generally accepted upper limit for lifetime of a compound nucleus.[19]
- ↑ This separation is based on that the resulting nuclei move past the target more slowly then the unreacted beam nuclei. The separator contains electric and magnetic fields whose effects on a moving particle cancel out for a specific velocity of a particle.[21] Such separation can also be aided by a time-of-flight measurement and a recoil energy measurement; a combination of the two may allow to estimate the mass of a nucleus.[22]
- ↑ Not all decay modes are caused by electrostatic repulsion. For example, beta decay is caused by the weak interaction.[29]
- ↑ It was already known by the 1960s that ground states of nuclei differed in energy and shape as well as that certain magic numbers of nucleons corresponded to greater stability of a nucleus. However, it was assumed that there was no nuclear structure in superheavy nuclei as they were too deformed to form one.[34]
- ↑ Since mass of a nucleus is not measured directly but is rather calculated from that of another nucleus, such measurement is called indirect. Direct measurements are also possible, but for the most part they have remained unavailable for superheavy nuclei.[39] The first direct measurement of mass of a superheavy nucleus was reported in 2018 at LBNL.[40] Mass was determined from the location of a nucleus after the transfer (the location helps determine its trajectory, which is linked to the mass-to-charge ratio of the nucleus, since the transfer was done in presence of a magnet).[41]
- ↑ If the decay occurred in a vacuum, then since total momentum of an isolated system before and after the decay must be preserved, the daughter nucleus would also receive a small velocity. The ratio of the two velocities, and accordingly the ratio of the kinetic energies, would thus be inverse to the ratio of the two masses. The decay energy equals the sum of the known kinetic energy of the alpha particle and that of the daughter nucleus (an exact fraction of the former).[30] The calculations hold for an experiment as well, but the difference is that the nucleus does not move after the decay because it is tied to the detector.
- ↑ Spontaneous fission was discovered by Soviet physicist Georgy Flerov,[42] a leading scientist at JINR, and thus it was a "hobbyhorse" for the facility.[43] In contrast, the LBL scientists believed fission information was not sufficient for a claim of synthesis of an element. They believed spontaneous fission had not been studied enough to use it for identification of a new element, since there was a difficulty of establishing that a compound nucleus had only ejected neutrons and not charged particles like protons or alpha particles.[19] They thus preferred to link new isotopes to the already known ones by successive alpha decays.[42]
- ↑ For instance, element 102 was mistakenly identified in 1957 at the Nobel Institute of Physics in Stockholm, Stockholm County, Sweden.[44] There were no earlier definitive claims of creation of this element, and the element was assigned a name by its Swedish, American, and British discoverers, nobelium. It was later shown that the identification was incorrect.[45] The following year, RL was unable to reproduce the Swedish results and announced instead their synthesis of the element; that claim was also disproved later.[45] JINR insisted that they were the first to create the element and suggested a name of their own for the new element, joliotium;[46] the Soviet name was also not accepted (JINR later referred to the naming of the element 102 as "hasty").[47] This name was proposed to IUPAC in a written response to their ruling on priority of discovery claims of elements, signed 29 September 1992.[47] The name "nobelium" remained unchanged on account of its widespread usage.[48]
References
- 1 2 Lide, David R., ed. (2003). CRC Handbook of Chemistry and Physics (84th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0484-9.
- 1 2 3 4 Fournier, Jean-Marc (1976). "Bonding and the electronic structure of the actinide metals". Journal of Physics and Chemistry of Solids. 37 (2): 235–244. Bibcode:1976JPCS...37..235F. doi:10.1016/0022-3697(76)90167-0.
- ↑ Dean, John A., ed. (1999). Lange's Handbook of Chemistry (15 ed.). McGraw-Hill. Section 4; Table 4.5, Electronegativities of the Elements.
- ↑ Sato, Tetsuya K.; Asai, Masato; Borschevsky, Anastasia; Beerwerth, Randolf; Kaneya, Yusuke; Makii, Hiroyuki; Mitsukai, Akina; Nagame, Yuichiro; Osa, Akihiko; Toyoshima, Atsushi; Tsukada, Kazuki; Sakama, Minoru; Takeda, Shinsaku; Ooe, Kazuhiro; Sato, Daisuke; Shigekawa, Yudai; Ichikawa, Shin-ichi; Düllmann, Christoph E.; Grund, Jessica; Renisch, Dennis; Kratz, Jens V.; Schädel, Matthias; Eliav, Ephraim; Kaldor, Uzi; Fritzsche, Stephan; Stora, Thierry (25 October 2018). "First Ionization Potentials of Fm, Md, No, and Lr: Verification of Filling-Up of 5f Electrons and Confirmation of the Actinide Series". Journal of the American Chemical Society. 140 (44): 14609–14613. doi:10.1021/jacs.8b09068.
- ↑ Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S.; Audi, G. (2021). "The NUBASE2020 evaluation of nuclear properties" (PDF). Chinese Physics C. 45 (3): 030001. doi:10.1088/1674-1137/abddae.
- ↑ Krämer, K. (2016). "Explainer: superheavy elements". Chemistry World. Retrieved 2020-03-15.
- ↑ "Discovery of Elements 113 and 115". Lawrence Livermore National Laboratory. Archived from the original on 2015-09-11. Retrieved 2020-03-15.
- ↑ Eliav, E.; Kaldor, U.; Borschevsky, A. (2018). "Electronic Structure of the Transactinide Atoms". In Scott, R. A. (ed.). Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley & Sons. pp. 1–16. doi:10.1002/9781119951438.eibc2632. ISBN 978-1-119-95143-8. S2CID 127060181.
- ↑ Oganessian, Yu. Ts.; Dmitriev, S. N.; Yeremin, A. V.; et al. (2009). "Attempt to produce the isotopes of element 108 in the fusion reaction 136Xe + 136Xe". Physical Review C. 79 (2): 024608. doi:10.1103/PhysRevC.79.024608. ISSN 0556-2813.
- ↑ Münzenberg, G.; Armbruster, P.; Folger, H.; et al. (1984). "The identification of element 108" (PDF). Zeitschrift für Physik A. 317 (2): 235–236. Bibcode:1984ZPhyA.317..235M. doi:10.1007/BF01421260. S2CID 123288075. Archived from the original (PDF) on 7 June 2015. Retrieved 20 October 2012.
- ↑ Subramanian, S. (28 August 2019). "Making New Elements Doesn't Pay. Just Ask This Berkeley Scientist". Bloomberg Businessweek. Retrieved 2020-01-18.
- 1 2 3 4 5 6 Ivanov, D. (2019). "Сверхтяжелые шаги в неизвестное" [Superheavy steps into the unknown]. nplus1.ru (in Russian). Retrieved 2020-02-02.
- ↑ Hinde, D. (2017). "Something new and superheavy at the periodic table". The Conversation. Retrieved 2020-01-30.
- ↑ Kern, B. D.; Thompson, W. E.; Ferguson, J. M. (1959). "Cross sections for some (n, p) and (n, α) reactions". Nuclear Physics. 10: 226–234. Bibcode:1959NucPh..10..226K. doi:10.1016/0029-5582(59)90211-1.
- ↑ Wakhle, A.; Simenel, C.; Hinde, D. J.; et al. (2015). Simenel, C.; Gomes, P. R. S.; Hinde, D. J.; et al. (eds.). "Comparing Experimental and Theoretical Quasifission Mass Angle Distributions". European Physical Journal Web of Conferences. 86: 00061. Bibcode:2015EPJWC..8600061W. doi:10.1051/epjconf/20158600061. hdl:1885/148847. ISSN 2100-014X.
- ↑ "Nuclear Reactions" (PDF). pp. 7–8. Retrieved 2020-01-27. Published as Loveland, W. D.; Morrissey, D. J.; Seaborg, G. T. (2005). "Nuclear Reactions". Modern Nuclear Chemistry. John Wiley & Sons, Inc. pp. 249–297. doi:10.1002/0471768626.ch10. ISBN 978-0-471-76862-3.
- 1 2 Krása, A. (2010). "Neutron Sources for ADS". Faculty of Nuclear Sciences and Physical Engineering. Czech Technical University in Prague: 4–8. S2CID 28796927.
- ↑ Wapstra, A. H. (1991). "Criteria that must be satisfied for the discovery of a new chemical element to be recognized" (PDF). Pure and Applied Chemistry. 63 (6): 883. doi:10.1351/pac199163060879. ISSN 1365-3075. S2CID 95737691.
- 1 2 Hyde, E. K.; Hoffman, D. C.; Keller, O. L. (1987). "A History and Analysis of the Discovery of Elements 104 and 105". Radiochimica Acta. 42 (2): 67–68. doi:10.1524/ract.1987.42.2.57. ISSN 2193-3405. S2CID 99193729.
- 1 2 3 4 Chemistry World (2016). "How to Make Superheavy Elements and Finish the Periodic Table [Video]". Scientific American. Retrieved 2020-01-27.
- ↑ Hoffman, Ghiorso & Seaborg 2000, p. 334.
- ↑ Hoffman, Ghiorso & Seaborg 2000, p. 335.
- ↑ Zagrebaev, Karpov & Greiner 2013, p. 3.
- ↑ Beiser 2003, p. 432.
- 1 2 Pauli, N. (2019). "Alpha decay" (PDF). Introductory Nuclear, Atomic and Molecular Physics (Nuclear Physics Part). Université libre de Bruxelles. Retrieved 2020-02-16.
- 1 2 3 4 5 Pauli, N. (2019). "Nuclear fission" (PDF). Introductory Nuclear, Atomic and Molecular Physics (Nuclear Physics Part). Université libre de Bruxelles. Retrieved 2020-02-16.
- ↑ Staszczak, A.; Baran, A.; Nazarewicz, W. (2013). "Spontaneous fission modes and lifetimes of superheavy elements in the nuclear density functional theory". Physical Review C. 87 (2): 024320–1. arXiv:1208.1215. Bibcode:2013PhRvC..87b4320S. doi:10.1103/physrevc.87.024320. ISSN 0556-2813.
- ↑ Audi et al. 2017, pp. 030001-129–030001-138.
- ↑ Beiser 2003, p. 439.
- 1 2 Beiser 2003, p. 433.
- ↑ Audi et al. 2017, p. 030001-125.
- ↑ Aksenov, N. V.; Steinegger, P.; Abdullin, F. Sh.; et al. (2017). "On the volatility of nihonium (Nh, Z = 113)". The European Physical Journal A. 53 (7): 158. Bibcode:2017EPJA...53..158A. doi:10.1140/epja/i2017-12348-8. ISSN 1434-6001. S2CID 125849923.
- ↑ Beiser 2003, p. 432–433.
- 1 2 3 Oganessian, Yu. (2012). "Nuclei in the "Island of Stability" of Superheavy Elements". Journal of Physics: Conference Series. 337 (1): 012005-1–012005-6. Bibcode:2012JPhCS.337a2005O. doi:10.1088/1742-6596/337/1/012005. ISSN 1742-6596.
- ↑ Moller, P.; Nix, J. R. (1994). Fission properties of the heaviest elements (PDF). Dai 2 Kai Hadoron Tataikei no Simulation Symposium, Tokai-mura, Ibaraki, Japan. University of North Texas. Retrieved 2020-02-16.
- 1 2 Oganessian, Yu. Ts. (2004). "Superheavy elements". Physics World. 17 (7): 25–29. doi:10.1088/2058-7058/17/7/31. Retrieved 2020-02-16.
- ↑ Schädel, M. (2015). "Chemistry of the superheavy elements". Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 373 (2037): 20140191. Bibcode:2015RSPTA.37340191S. doi:10.1098/rsta.2014.0191. ISSN 1364-503X. PMID 25666065.
- ↑ Hulet, E. K. (1989). Biomodal spontaneous fission. 50th Anniversary of Nuclear Fission, Leningrad, USSR. Bibcode:1989nufi.rept...16H.
- ↑ Oganessian, Yu. Ts.; Rykaczewski, K. P. (2015). "A beachhead on the island of stability". Physics Today. 68 (8): 32–38. Bibcode:2015PhT....68h..32O. doi:10.1063/PT.3.2880. ISSN 0031-9228. OSTI 1337838. S2CID 119531411.
- ↑ Grant, A. (2018). "Weighing the heaviest elements". Physics Today. doi:10.1063/PT.6.1.20181113a. S2CID 239775403.
- ↑ Howes, L. (2019). "Exploring the superheavy elements at the end of the periodic table". Chemical & Engineering News. Retrieved 2020-01-27.
- 1 2 Robinson, A. E. (2019). "The Transfermium Wars: Scientific Brawling and Name-Calling during the Cold War". Distillations. Retrieved 2020-02-22.
- ↑ "Популярная библиотека химических элементов. Сиборгий (экавольфрам)" [Popular library of chemical elements. Seaborgium (eka-tungsten)]. n-t.ru (in Russian). Retrieved 2020-01-07. Reprinted from "Экавольфрам" [Eka-tungsten]. Популярная библиотека химических элементов. Серебро — Нильсборий и далее [Popular library of chemical elements. Silver through nielsbohrium and beyond] (in Russian). Nauka. 1977.
- ↑ "Nobelium - Element information, properties and uses | Periodic Table". Royal Society of Chemistry. Retrieved 2020-03-01.
- 1 2 Kragh 2018, pp. 38–39.
- ↑ Kragh 2018, p. 40.
- 1 2 Ghiorso, A.; Seaborg, G. T.; Oganessian, Yu. Ts.; et al. (1993). "Responses on the report 'Discovery of the Transfermium elements' followed by reply to the responses by Transfermium Working Group" (PDF). Pure and Applied Chemistry. 65 (8): 1815–1824. doi:10.1351/pac199365081815. S2CID 95069384. Archived (PDF) from the original on 25 November 2013. Retrieved 7 September 2016.
- ↑ Commission on Nomenclature of Inorganic Chemistry (1997). "Names and symbols of transfermium elements (IUPAC Recommendations 1997)" (PDF). Pure and Applied Chemistry. 69 (12): 2471–2474. doi:10.1351/pac199769122471.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Barber, Robert C.; Greenwood, Norman N.; Hrynkiewicz, Andrzej Z.; Jeannin, Yves P.; Lefort, Marc; Sakai, Mitsuo; Úlehla, Ivan M.; Wapstra, Aaldert Hendrik; Wilkinson, Denys H. (1993). "Discovery of the transfermium elements. Part II: Introduction to discovery profiles. Part III: Discovery profiles of the transfermium elements". Pure and Applied Chemistry. 65 (8): 1757. doi:10.1351/pac199365081757. S2CID 195819585. (Note: for Part I see Pure and Applied Chemistry, vol. 63, no. 6, pp. 879–886, 1991)
- 1 2 Silva 2011, pp. 1636–7
- ↑ Fields, Peter R.; Friedman, Arnold M.; Milsted, John; Atterling, Hugo; Forsling, Wilhelm; Holm, Lennart W.; Åström, Björn (1 September 1957). "Production of the New Element 102". Physical Review. 107 (5): 1460–1462. Bibcode:1957PhRv..107.1460F. doi:10.1103/PhysRev.107.1460.
- 1 2 3 4 5 6 Emsley, John (2011). Nature's Building Blocks: An A-Z Guide to the Elements. Oxford University Press. pp. 368–9. ISBN 978-0-19-960563-7.
- 1 2 Ghiorso, Albert; Seaborg, Glenn T.; Oganessian, Yuri Ts.; Zvara, Ivo; Armbruster, Peter; Hessberger, F. P.; Hofmann, Sigurd; Leino, Matti E.; Münzenberg, Gottfried; Reisdorf, Willibrord; Schmidt, Karl-Heinz (1993). "Responses on 'Discovery of the transfermium elements' by Lawrence Berkeley Laboratory, California; Joint Institute for Nuclear Research, Dubna; and Gesellschaft fur Schwerionenforschung, Darmstadt followed by reply to responses by the Transfermium Working Group". Pure and Applied Chemistry. 65 (8): 1815–1824. doi:10.1351/pac199365081815.
- ↑ Karpenko, V. (1980). "The Discovery of Supposed New Elements: Two Centuries of Errors". Ambix. 27 (2): 77–102. doi:10.1179/amb.1980.27.2.77.
- 1 2 "Names and symbols of transfermium elements" (PDF). Pure and Applied Chemistry. 69 (12): 2471–2473. 1997. doi:10.1351/pac199769122471.
- ↑ Hoffmann, Darleane C.; Lee, Diana M.; Pershina, Valeria (2006). "Transactinides and the future elements". In Morss, Lester R.; Edelstein, Norman M.; Fuger, Jean (eds.). The Chemistry of the Actinide and Transactinide Elements (3rd ed.). Springer. p. 1660. ISBN 978-1-4020-3555-5.
- ↑ "Element 114 is Named Flerovium and Element 116 is Named Livermorium" (Press release). IUPAC. 30 May 2012. Archived from the original on 2 June 2012.
- ↑ Haire, Richard G. (2006). "Einsteinium". In Morss, Lester R.; Edelstein, Norman M.; Fuger, Jean (eds.). The Chemistry of the Actinide and Transactinide Elements (PDF). Vol. 3 (3rd ed.). Dordrecht, the Netherlands: Springer. pp. 1577–1620. doi:10.1007/1-4020-3598-5_12. ISBN 978-1-4020-3555-5. Archived from the original (PDF) on 2010-07-17. Retrieved 2014-08-15.
- 1 2 3 4 5 6 Silva 2011, p. 1639
- 1 2 Silva 2011, pp. 1626–8
- ↑ Johansson, Börje; Rosengren, Anders (1975). "Generalized phase diagram for the rare-earth elements: Calculations and correlations of bulk properties". Physical Review B. 11 (8): 2836–2857. Bibcode:1975PhRvB..11.2836J. doi:10.1103/PhysRevB.11.2836.
- ↑ Hulet, E. Kenneth (1980). "Chapter 12. Chemistry of the Heaviest Actinides: Fermium, Mendelevium, Nobelium, and Lawrencium". In Edelstein, Norman M. (ed.). Lanthanide and Actinide Chemistry and Spectroscopy. ACS Symposium Series. Vol. 131. pp. 239–263. doi:10.1021/bk-1980-0131.ch012. ISBN 978-0-8412-0568-0.
- ↑ Haynes, William M., ed. (2011). CRC Handbook of Chemistry and Physics (92nd ed.). CRC Press. pp. 4.121–4.123. ISBN 978-1-4398-5511-9.
- 1 2 3 4 5 6 7 8 9 10 11 12 Silva 2011, pp. 1639–41
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. p. 1278. ISBN 978-0-08-037941-8.
- ↑ Balasubramanian, Krishnan (4 December 2001). "Potential energy surfaces of Lawrencium and Nobelium dihydrides (LrH2 and NoH2)…". Journal of Chemical Physics. 116 (9): 3568–75. Bibcode:2002JChPh.116.3568B. doi:10.1063/1.1446029.
- ↑ Toyoshima, A.; Kasamatsu, Y.; Tsukada, K.; Asai, M.; Kitatsuji, Y.; Ishii, Y.; Toume, H.; Nishinaka, I.; Haba, H.; Ooe, K.; Sato, W.; Shinohara, A.; Akiyama, K.; Nagame, Y. (8 July 2009). "Oxidation of element 102, nobelium, with flow electrolytic column chromatography on an atom-at-a-time scale". Journal of the American Chemical Society. 131 (26): 9180–1. doi:10.1021/ja9030038. PMID 19514720.
- ↑ Martin, William C.; Hagan, Lucy; Reader, Joseph; Sugar, Jack (1974). "Ground Levels and Ionization Potentials for Lanthanide and Actinide Atoms and Ions" (PDF). Journal of Physical and Chemical Reference Data. 3 (3): 771–9. Bibcode:1974JPCRD...3..771M. doi:10.1063/1.3253147. S2CID 97945150. Archived from the original (PDF) on 2020-02-15.
- ↑ Lide, David R. (editor), CRC Handbook of Chemistry and Physics, 84th Edition, CRC Press, Boca Raton (FL), 2003, section 10, Atomic, Molecular, and Optical Physics; Ionization Potentials of Atoms and Atomic Ions
- 1 2 3 4 5 6 7 8 Silva 2011, pp. 1637–8
- ↑ Joint Institute for Nuclear Research, 2020 (PDF) (Report). 23 June 2021. pp. 117–118. Retrieved 26 June 2021.
- 1 2 3 4 5 6 7 "Nucleonica :: Web driven nuclear science".
- 1 2 3 4 5 6 7 Audi, Georges; Bersillon, Olivier; Blachot, Jean; Wapstra, Aaldert Hendrik (2003), "The NUBASE evaluation of nuclear and decay properties", Nuclear Physics A, 729: 3–128, Bibcode:2003NuPhA.729....3A, doi:10.1016/j.nuclphysa.2003.11.001
- ↑ Kratz, Jens Volker (5 September 2011). The Impact of Superheavy Elements on the Chemical and Physical Sciences (PDF). 4th International Conference on the Chemistry and Physics of the Transactinide Elements. Retrieved 27 August 2013.
- ↑ Nurmia, Matti (2003). "Nobelium". Chemical and Engineering News. 81 (36): 178. doi:10.1021/cen-v081n036.p178.
- 1 2 3 4 Silva 2011, pp. 1638–9
Bibliography
- Audi, G.; Kondev, F. G.; Wang, M.; et al. (2017). "The NUBASE2016 evaluation of nuclear properties" (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- Beiser, A. (2003). Concepts of modern physics (6th ed.). McGraw-Hill. ISBN 978-0-07-244848-1. OCLC 48965418.
- Hoffman, D. C.; Ghiorso, A.; Seaborg, G. T. (2000). The Transuranium People: The Inside Story. World Scientific. ISBN 978-1-78-326244-1.
- Silva, Robert J. (2011). "Chapter 13. Fermium, Mendelevium, Nobelium, and Lawrencium". In Morss, Lester R.; Edelstein, Norman M.; Fuger, Jean (eds.). The Chemistry of the Actinide and Transactinide Elements. Netherlands: Springer. pp. 1621–1651. doi:10.1007/978-94-007-0211-0_13. ISBN 978-94-007-0210-3.
- Zagrebaev, V.; Karpov, A.; Greiner, W. (2013). "Future of superheavy element research: Which nuclei could be synthesized within the next few years?". Journal of Physics: Conference Series. 420 (1): 012001. arXiv:1207.5700. Bibcode:2013JPhCS.420a2001Z. doi:10.1088/1742-6596/420/1/012001. ISSN 1742-6588. S2CID 55434734.
External links
- Chart of Nuclides Archived 2018-10-10 at the Wayback Machine. nndc.bnl.gov
- Los Alamos National Laboratory – Nobelium
- Nobelium at The Periodic Table of Videos (University of Nottingham)